E-Submission
E-SubmissionPubMed Central, CAS, DOAJ, KCI

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 40(2); 2023 > Article
-
Review article
Hepatic ischemia-reperfusion injury with respect to oxidative stress and inflammatory response: a narrative review -
Eun Kyung Choi1
 , Dong Gun Lim2
, Dong Gun Lim2 -
Journal of Yeungnam Medical Science 2023;40(2):115-122.
DOI: https://doi.org/10.12701/jyms.2022.00017
Published online: March 21, 2022
1Department of Anesthesiology and Pain Medicine, Yeungnam University College of Medicine, Daegu, Korea
2Department of Anesthesiology and Pain Medicine, School of Medicine, Kyungpook National University, Daegu, Korea
- Correspondence author: Dong Gun Lim, MD, PhD Department of Anesthesiology and Pain Medicine, School of Medicine, Kyungpook National University, 130 Dongdeok-ro, Jung-gu, Daegu 41944, Korea Tel: +82-53-420-5876 • Fax: +82-53-426-2760 • E-mail: dglim@knu.ac.kr
Copyright © 2023 Yeungnam University College of Medicine, Yeungnam University Institute of Medical Science
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Hepatic ischemia-reperfusion injury is a major complication of liver transplantation, trauma, and shock. This pathological condition can lead to graft dysfunction and rejection in the field of liver transplantation and clinical hepatic dysfunction with increased mortality. Although the pathological mechanisms of hepatic ischemia-reperfusion injury are very complex, and several intermediators and cells are involved in this phenomenon, oxidative stress and inflammatory responses are the key processes that aggravate hepatic injury. This review summarizes the current understanding of oxidative stress and inflammatory responses and, in that respect, addresses the therapeutic approaches to attenuate hepatic ischemia-reperfusion injury.
- Ischemia-reperfusion injury (IRI) is characterized by initial organ underperfusion (ischemia), followed by restoration of blood flow (reperfusion) [1]. Although restoration of oxygen delivery to an ischemic organ is needed to prevent hypoxic cellular damage, reperfusion may accentuate organ injury in excess of the stress produced by ischemia itself [1]. IRI can occur in diverse clinical settings including organ transplantation, trauma, shock, cardiopulmonary bypass, and thrombolytic therapy. Hepatic IRI is a major complication of hepatic resection surgery (e.g., the Pringle maneuver) and liver transplantation. This pathological condition can lead to liver cellular damage and clinical hepatic dysfunction, and may even predispose to distant organ failure.
Introduction
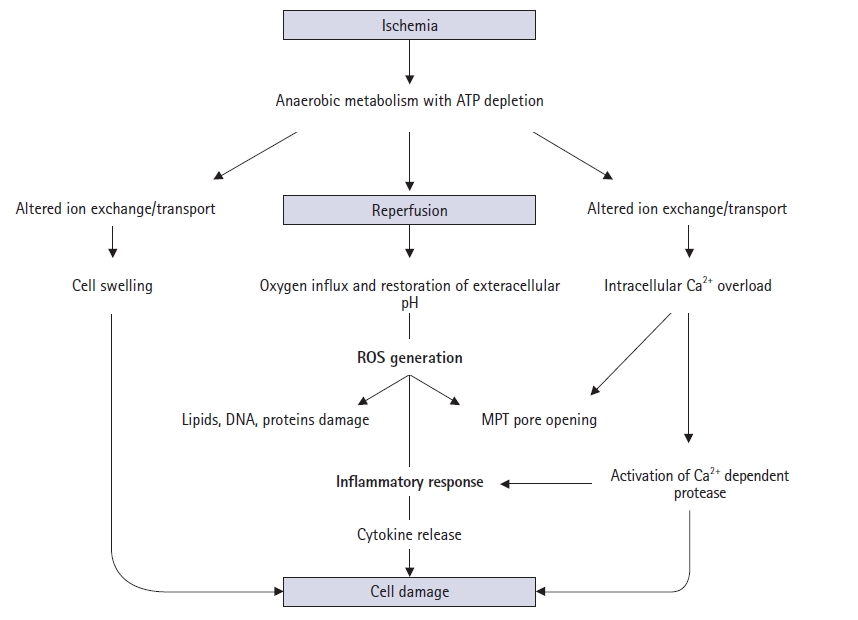
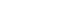
- Various pathophysiological mechanisms have been proposed for hepatic IRI, but the actual mechanisms remain unclear. Hepatic IRI occurs in two main settings. First, ischemia can follow temporary vascular occlusion of the hepatic pedicle or various forms of shock and trauma, whereby hypoxic injury occurs. Second, reperfusion injury can be added to hepatic ischemic injury. This phenomenon is a dynamic process that leads to metabolic acidosis, intracellular calcium overload, mitochondrial damage, Kupffer cell activation, oxidative stress, inflammatory responses, and necrotic or apoptotic cell death (Fig. 1) [2].
- 1. Metabolic acidosis
- Metabolic acidosis is the basic mechanism underlying hepatic IRI [3]. It results from anaerobic glycolysis during ischemia, which leads to depletion of adenosine 5′-triphosphate (ATP) from organs, consequently producing lactate. During reperfusion, the tissue pH increases, leading to the activation of phospholipases and proteolytic enzymes, which in turn cause cell damage, necrosis, and apoptosis, resulting in IRI [3].
- 2. Intracellular calcium overload
- Intracellular calcium homeostasis is maintained by the Na+/K+ and H+/Ca2+ exchange systems. During ischemia and reperfusion, ATP depletion leads to a decrease in ATP-dependent Na+/K+ ATPase activity in the cell membrane. This results in increased intracellular Na+ concentrations, leading to the inward flux of calcium ions [4]. In addition, increased ischemia-induced permeability of cell membranes causes further movement of calcium ions into the cell, and a large number of calcium ions are released from the endoplasmic reticulum and damaged mitochondria. Intracellular calcium overload occurs, which in turn interferes with cellular metabolic pathways [5].
- 3. Mitochondrial damage
- Mitochondria act as pathological triggers, mediators, and effectors of hepatic IRI [6]. Mitochondrial functions normally involve several processes, including energy production, cell survival, and programmed cell death [7]. However, the dysfunction in pathological ischemia and reperfusion is initiated by mitochondrial permeability transition (MPT) pore opening [8]. ATP depletion, calcium ion overload, and toxic oxidant release promote MPT onset, which follows depolarization of the mitochondrial membrane potential, matrix swelling, and membrane rupture [9]. Moreover, MPT opening can lead to apoptosis by mitochondrial swelling and the subsequent release of cytochrome C [10].
- 4. Oxidative stress
- Reactive oxygen species (ROS) normally exist as by-products of cellular metabolism in proteins, lipids, nucleic acids, and other biologically active molecules. However, the nature and amount of ROS change during IRI. Aerobic cells use molecular oxygen to remove electrons during oxidative catabolism (from O2 to H2O) in the mitochondrial respiratory chain. However, small amounts of oxygen (1%–3%) are reduced through the univalent pathway, forming reactive intermediate species, including superoxide anion (O2•–), hydrogen peroxide (H2O2), and hydroxyl radical (•HO) [11,12]. Metal ions such as iron and copper react with hydrogen peroxide via the Fenton reaction, producing the toxic hydroxyl radical [13]. Superoxide anion and these reactive intermediates are known as ROS [12].
- Many metabolic processes, including the enzymatic activities of xanthine oxidase (XO) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, for example, and mitochondrial respiration produce large amounts of ROS [14]. Although XO is known to be an important mediator of ROS formation, mitochondria have recently been suggested as the main production site for large amounts of superoxide, leading to the formation of MPT pores that can cause cell death [15].
- Nitric oxide (NO), nitrogen dioxide, and peroxynitrite (ONOO–) are biologically important reactive nitrogen species (RNS), the last two of which result from the interaction of NO with molecular oxygen [16]. Among these, ONOO–, a strong oxidizing agent generated from superoxide anion and NO, can attack basic cell constituents, such as DNA and proteins [17]. NO, a gaseous signaling molecule is produced by the enzymatically catalyzed reaction between L-arginine and oxygen [18]. During hepatic IRI, two main NO synthases (NOS), endothelial (eNOS) and inducible (iNOS), synthesize NO, which can either prevent or promote cell injury [19]. eNOS is constitutively expressed in sinusoidal endothelial cells, whereas iNOS is stimulated by numerous cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) [19]. NO exerts a protective effect on parenchymal hepatocytes by preventing the action of TNF-α and apoptotic factors, blocking MPT onset, and preventing sinusoidal obstruction by inducing vasodilatation, neutrophil accumulation, and platelet adhesion [20-22]. However, overproduction of NO during the late reperfusion period can result in high levels of injurious ROS and the accumulation of inflammatory cytokines by increased iNOS expression and decreased eNOS expression [18]. In addition, NO can be converted into the toxic ONOO–, which can cause tissue injury through multiple pathways, including lipid peroxidation, inhibition of the mitochondrial respiratory chain, and modification of protein nitrotyrosine levels [23-25]. Thus, NO can promote or prevent cell survival, depending on its concentration, activation time, and NO-superoxide radical ratio.
- Therefore, ROS and RNS directly react with numerous biological molecules, leading to tissue toxicity. The above effects damage sinusoidal endothelial cells, increasing permeability of the microvasculature and promoting neutrophil and platelet adhesion to these cells, followed by subsequent disruption of the microcirculation [26]. Moreover, these oxygen radicals lead to hepatocellular apoptosis by influencing intracellular signaling pathways via effects on gene expression and direct oxidation of nuclear DNA structure in the hepatic parenchyma [27].
- In contrast to the generation of ROS and RNS, the presence of endogenous antioxidant enzymes attenuates further hepatic injury. When present at low concentrations, antioxidant enzymes can prevent oxidative damage and detoxify ROS [28]. Hepatocytes contain high levels of intracellular antioxidant enzymes, including superoxide dismutase (SOD), glutathione peroxidase, and catalase; however, during IRI, an imbalance between ROS and endogenous antioxidant enzymes occurs, consequently leading to damage to nucleic acids, proteins, and lipids [27]. SOD catalyzes the dismutation of superoxide anion to hydrogen peroxide and oxygen [27]. Hydrogen peroxide can be decomposed via three main systems. First, catalase breaks down hydrogen peroxide into oxygen and water [27]. Second, glutathione peroxidase removes hydrogen peroxide via glutathione oxidation to glutathione disulfide [27]. Finally, peroxiredoxins reduce hydrogen peroxide to water [29].
- 5. Inflammatory responses
- Hepatic IRI is characterized by inflammatory responses in the postischemic tissue. During ischemia, a lack of ATP causes failure of the Na+/K+ ATPase and subsequent intracellular Na+ accumulation with cellular swelling in hepatocytes, Kupffer cells, and sinusoidal endothelial cells. Here, increased endothelin and decreased NO (a vasoconstrictor and vasodilator, respectively) levels induce cellular swelling, which in turn leads to sinusoidal narrowing [30]. During reperfusion, the attachment of neutrophils and platelets to the sinusoid with increased adhesion molecules leads to defects in hepatic microcirculation and even the complete absence of blood flow and reflow [26].
- Kupffer cells, the resident hepatic macrophages, play a pivotal role in initiating hepatic cellular damage in IRI [31]. During ischemia and the early reperfusion period, Kupffer cells release proinflammatory mediators, such as TNF-α, IL-1, platelet-activating factor, and ROS, which activate a cascade of inflammatory responses [32]. These inflammatory cytokines, chemokines, and small molecule mediators recruit neutrophils and induce ROS production and further inflammation, exacerbating tissue damage during the late reperfusion period [33]. In addition, Kupffer cells activate CD4+ T lymphocytes in the early reperfusion period, preceding neutrophil accumulation induced by the chemotactic agent IL-17. Reciprocally, CD4+ T cells release interferon-gamma, which activates Kupffer cells to generate TNF-α and IL-1 [34,35]. Over a time scale similar to that of the CD4+ T cells, natural killer cells, another leukocyte subset, are recruited to the liver; they produce interferon-gamma, aggravating IRI [36].
- The complement system and cytokines are important humoral factors involved in hepatic IRI. Once activated in IRI, complement can damage either directly by lysing hepatocytes through the membrane attack complex or indirectly by activating Kupffer cells and neutrophils [26]. Among the complement components, C5a is the most potent inflammatory mediator that releases proinflammatory cytokines, including TNF-α, IL-1, and IL-6 [37]. In addition, C5a inhibits endothelium-dependent relaxation and alters vascular tone, which further compromises the blood flow to ischemic tissues [37].
- Numerous cytokines can play one of two roles, either proinflammatory or anti-inflammatory. TNF-α is a crucial proinflammatory cytokine in the hepatic inflammatory response during ischemia and reperfusion. Although various cells in the liver release TNF-α, its production by Kupffer cells is the most prominent [38]. Upregulation of TNF-α during ischemia and reperfusion results in ROS activation, expression of various adhesion molecules such as intercellular adhesion molecule 1 and P-selectin, and thus recruitment of neutrophils into the liver [39]. Similarly, IL-1 can induce ROS production and promote leukocyte aggregation [40]. Conversely, IL-6 produced by Kupffer cells has a protective effect that is mediated by the downregulation of oxidative stress markers and increase in glutathione, an antioxidant, thus reducing hepatocyte damage [41].
- One question that arises is how immune cells are stimulated by pathogens in surgical settings. The answer begins with hepatic oxidative stress. During ischemia and reperfusion, ROS and RNS generated by mitochondrial respiration threaten hepatocyte viability [42]. Damaged hepatocytes and other immune cells (e.g., Kupffer cells and neutrophils) release pathogenic endogenous molecules and danger-associated molecular patterns (DAMPs) that overactivate innate immune responses [43]. DAMPs and self-antigens are normally physiological constituents of healthy cells; however, they become immunostimulators in the extracellular environment. Consequently, DAMPs stimulate Kupffer cells, which results in the production of inflammatory mediators, such as cytokines, chemokines, and ROS. This process induces reperfusion injury via intense neutrophil infiltration [32]. In other words, oxygen-free radicals and proinflammatory cytokines released by activated Kupffer cells can promote the infiltration of neutrophils and platelets into sinusoidal endothelium, thereby disrupting hepatic microcirculation and further aggravating hepatic injury [44]. Among the various DAMPs in hepatic IRI, high-mobility group box-1 is the best characterized. It is released by damaged hepatocytes and interacts with toll-like receptors (TLRs), particularly TLR-4 [45]. In this instance, several signaling transcription factors mediate TLR-4 activation, including nuclear factor-kappa B, activating protein-1, and mitogen-activated protein kinases (ERK, JNK, and P38), which modulate gene expression correlated with inflammatory progression [45,46]. Thus, DAMP-derived danger signals mediate the contribution of leukocytes to the severity of liver damage-induced ischemia and reperfusion.
Pathophysiology
1) Reactive oxygen species
2) Reactive nitrogen species
3) Antioxidant systems
1) Inflammatory cells
2) Complement and cytokines
3) Endogenous danger signals
- 1. Modulation of oxidative stress
- Oxidative stress occurs when oxidants are overproduced or antioxidant levels are reduced. Therefore, treatment strategies for oxidant modulation include the inhibition of ROS formation, scavenging of ROS, and potentiation of endogenous antioxidant capacity. As mentioned above, XO, NADPH oxidase, and MPT collectively contribute to ROS formation. Many studies have demonstrated the protective effects of inhibition of these enzymes in hepatic IRI. For example, known inhibitors are allopurinol [47] and apocynin [48] for XO and NADPH oxidase, respectively. Additionally, edaravone, a mitochondria-specific antioxidant with protective effects against hepatic IRI, has been experimentally confirmed as an MPT inhibitor. It exerts its effect by blocking the MPT and maintaining an adequate ATP concentration [49]. Moreover, cyclosporin A inhibits MPT pore opening in the mitochondrial matrix; however, its clinical use remains limited [50].
- Normally, the antioxidant defense system controls ROS production. Various antioxidant defenses have been demonstrated to have beneficial effects, both experimentally and clinically, in hepatic IRI. Antioxidants are a heterogeneous family of molecules that can be classified according to their site of action as follows: intracellular, membrane, and extracellular. Representative intracellular antioxidant enzymes are SOD, glutathione peroxidase, and catalase [51-53]. Alpha-tocopherol and coenzyme Q are the main membrane antioxidants [54], whereas metal-binding proteins, such as transferrin and ceruloplasmin, are major extracellular antioxidants that sequester free iron and copper ions that can promote oxidative damage, respectively [55]. In addition, many low-molecular-weight substances that are synthesized in vivo [56] (e.g., melatonin, coenzyme Q, and uric acid) or dietary constituents (e.g., vitamins C and E) exert antioxidant properties [57,58]. These antioxidants have a systematic relationship in the antioxidant network, and they counteract and exhibit synergism [59].
- Among the many interventions against oxidative stress, ischemic preconditioning has been shown to have beneficial effects against hepatic IRI. This preconditioning requires pre-exposure of the liver to brief ischemic episodes to increase its tolerance against subsequent detrimental insults [60]. The underlying molecular mechanism of this intervention is that mild burst oxidants, especially hydrogen peroxide, generated during ischemic preconditioning trigger specific biochemical pathways that ultimately protect against further oxidative damage and lead to adaptation [61]. However, the clinical implications of ischemic preconditioning may be limited because of its invasive properties. Remote ischemic preconditioning is less invasive and more clinically relevant [62]. Remote ischemic preconditioning comprises signal generation from remote organs, signal transfer to target organs, and subsequent protective effects in the target organs. Various neural and humoral factors, such as autonomic ganglion, bradykinin, and adenosine, have been implicated in the pathophysiologic mechanisms of remote ischemic preconditioning [62]. A practical technique has been proposed, which encompasses several brief episodes of ischemia and reperfusion in a remote organ that protects distant targets.
- 2. Modulation of inflammatory response
- As previously mentioned, activation of the immune system is a crucial factor in hepatic IRI, and Kupffer cells and chemoattracted neutrophils are important culprits. Consequently, the cytokine network connected to DAMP contributes to the severity of hepatic IRI via a wave of ROS generation. Consistent with this finding, anti-inflammatory therapy, through various biochemical intersections that inhibit the inflammatory cascade, can attenuate leukocyte recruitment and ROS generation. First, inhibition of DAMP can prevent inflammation and oxidative stress [63]. In addition, administration of a mitochondria-selective S-nitrosylating agent during the acute reperfusion period could prevent mitochondrial ROS bursts and the resulting DAMP release [64]. Further downstream, direct inhibition of Kupffer cells and neutrophils is a promising strategy to treat hepatic IRI [65,66]. In liver transplantation, Kupffer cells are primed in cold ischemic solutions. Subsequently, the primed Kupffer cells exhibit progressive rounding, vacuolization, and degranulation [67]; hence, modulation of Kupffer cell activation plays an important role in reducing IRI in liver transplantation. In contrast, therapeutic modalities to prevent neutrophil recruitment are more diverse due to their multistep processes: chemokine production, expression of adhesion molecules to attach to endothelial cells, and release of effector molecules such as ROS [68]. In a similar context, inhibition of inflammatory cytokines (e.g., TNF-α and IL-1) could be a worthwhile treatment option [38,67]. As TNF-α is a key inflammatory mediator, its neutralization with an antibody and inhibition of its production attenuate hepatic IRI involving neutrophil infiltration [38,69]. Anti-inflammation reduces oxidative stress; conversely, inhibition of ROS and RNS is also a potential therapeutic method for relieving inflammation because these oxidants can activate Kupffer cells and neutrophils, followed by a second wave of ROS and RNS generation. Moreover, inhibition of the complement cascade attenuates hepatic injury [70].
Protective strategies for hepatic ischemia-reperfusion injury
1) Ischemic preconditioning
- Hepatic IRI occurs in various clinical settings and is a major cause of morbidity and mortality. Although numerous interactions and mediators are involved in its pathophysiology, oxidative stress and inflammatory responses are the main mechanisms. Several therapeutic methods that limit oxidative stress and inflammatory responses have been suggested and applied to attenuate hepatic IRI. If based on a basic understanding of the aforementioned main pathological mechanisms and therapeutic modalities that could improve patient care, our knowledge of these complex hepatic IRI mechanisms remains incomplete.
Conclusion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Funding
None.
-
Author contributions
Conceptualization: EKC, DGL; Data curation, Methodology: EKC; Formal analysis, Supervision, Validation: DGL; Writing-original draft: EKC; Writing-review & editing: EKC, DGL
Notes
- 1. Mendes-Braz M, Elias-Miró M, Jiménez-Castro MB, Casillas-Ramírez A, Ramalho FS, Peralta C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J Biomed Biotechnol 2012;2012:298657.ArticlePubMedPMC
- 2. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012;298:229–317.ArticlePubMedPMC
- 3. Heijnen BH, Elkhaloufi Y, Straatsburg IH, Van Gulik TM. Influence of acidosis and hypoxia on liver ischemia and reperfusion injury in an in vivo rat model. J Appl Physiol (1985) 2002;93:319–23.ArticlePubMed
- 4. Li J, Li RJ, Lv GY, Liu HQ. The mechanisms and strategies to protect from hepatic ischemia-reperfusion injury. Eur Rev Med Pharmacol Sci 2015;19:2036–47.PubMed
- 5. Gnaiger E, Kuznetsov AV, Rieger G, Amberger A, Fuchs A, Stadlmann S, et al. Mitochondrial defects by intracellular calcium overload versus endothelial cold ischemia/reperfusion injury. Transpl Int 2000;13(Suppl 1):S555–7.ArticlePubMed
- 6. Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol 2011;2:13.ArticlePubMedPMC
- 7. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004;287:C817-33.
- 8. Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol 1997;273:C1783-92.
- 9. Stowe DF, Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal 2009;11:1373–414.ArticlePubMedPMC
- 10. Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr Mol Med 2003;3:527–35.ArticlePubMed
- 11. Nohl H, Gille L, Kozlov A, Staniek K. Are mitochondria a spontaneous and permanent source of reactive oxygen species? Redox Rep 2003;8:135–41.ArticlePubMed
- 12. Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys 1992;298:446–51.ArticlePubMed
- 13. Sutton HC, Winterbourn CC. On the participation of higher oxidation states of iron and copper in Fenton reactions. Free Radic Biol Med 1989;6:53–60.ArticlePubMed
- 14. Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med (Berl) 1999;77:577–92.ArticlePubMed
- 15. Nieminen AL, Byrne AM, Herman B, Lemasters JJ. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am J Physiol 1997;272(4 Pt 1):C1286–94.ArticlePubMed
- 16. Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med 2007;43:645–57.ArticlePubMedPMC
- 17. Mitrogianni Z, Barbouti A, Galaris D, Siamopoulos KC. Tyrosine nitration in plasma proteins from patients undergoing hemodialysis. Am J Kidney Dis 2004;44:286–92.ArticlePubMed
- 18. Fondevila C, Shen XD, Tsuchihashi S, Uchida Y, Freitas MC, Ke B, et al. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury. Liver Transpl 2008;14:1133–41.ArticlePubMedPMC
- 19. Chiang N, Gronert K, Clish CB, O’Brien JA, Freeman MW, Serhan CN. Leukotriene B4 receptor transgenic mice reveal novel protective roles for lipoxins and aspirin-triggered lipoxins in reperfusion. J Clin Invest 1999;104:309–16.ArticlePubMedPMC
- 20. Inderbitzin D, Beldi G, Avital I, Vinci G, Candinas D. Local and remote ischemia-reperfusion injury is mitigated in mice overexpressing human C1 inhibitor. Eur Surg Res 2004;36:142–7.ArticlePubMed
- 21. Peralta C, Fernández L, Panés J, Prats N, Sans M, Piqué JM, et al. Preconditioning protects against systemic disorders associated with hepatic ischemia-reperfusion through blockade of tumor necrosis factor-induced P-selectin up-regulation in the rat. Hepatology 2001;33:100–13.ArticlePubMed
- 22. Rajesh M, Pan H, Mukhopadhyay P, Bátkai S, Osei-Hyiaman D, Haskó G, et al. Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J Leukoc Biol 2007;82:1382–9.ArticlePubMed
- 23. Doulias PT, Barbouti A, Galaris D, Ischiropoulos H. SIN-1-induced DNA damage in isolated human peripheral blood lymphocytes as assessed by single cell gel electrophoresis (comet assay). Free Radic Biol Med 2001;30:679–85.ArticlePubMed
- 24. Hon WM, Lee KH, Khoo HE. Nitric oxide in liver diseases: friend, foe, or just passerby? Ann N Y Acad Sci 2002;962:275–95.ArticlePubMed
- 25. Szabó C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 2003;140-1:105–12.ArticlePubMed
- 26. Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res 2008;147:153–9.ArticlePubMed
- 27. Haddad JJ. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal 2002;14:879–97.ArticlePubMed
- 28. Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995;41(12 Pt 2):1819–28.ArticlePubMed
- 29. Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem 1998;273:6297–302.ArticlePubMed
- 30. Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric oxide mechanism of protection in ischemia and reperfusion injury. J Invest Surg 2009;22:46–55.ArticlePubMed
- 31. Caban A, Oczkowicz G, Abdel-Samad O, Cierpka L. Influence of Kupffer cells on the early phase of liver reperfusion. Transplant Proc 2002;34:694–7.ArticlePubMed
- 32. Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol 1991;260(3 Pt 1):G355–62.ArticlePubMed
- 33. Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury. J Gastroenterol Hepatol 2000;15:718–24.ArticlePubMed
- 34. Hanschen M, Zahler S, Krombach F, Khandoga A. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation 2008;86:710–8.ArticlePubMed
- 35. Caldwell CC, Tschoep J, Lentsch AB. Lymphocyte function during hepatic ischemia/reperfusion injury. J Leukoc Biol 2007;82:457–64.ArticlePubMed
- 36. van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev 2012;23:69–84.ArticlePubMed
- 37. Collard CD, Lekowski R, Jordan JE, Agah A, Stahl GL. Complement activation following oxidative stress. Mol Immunol 1999;36:941–8.ArticlePubMed
- 38. Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA Jr. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest 1990;85:1936–43.ArticlePubMedPMC
- 39. Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 2006;290:G583–9.ArticlePubMed
- 40. Kato A, Gabay C, Okaya T, Lentsch AB. Specific role of interleukin-1 in hepatic neutrophil recruitment after ischemia/reperfusion. Am J Pathol 2002;161:1797–803.ArticlePubMedPMC
- 41. Husted TL, Blanchard J, Schuster R, Shen H, Lentsch AB. Potential role for IL-23 in hepatic ischemia/reperfusion injury. Inflamm Res 2006;55:177–8.ArticlePubMed
- 42. Mukhopadhyay P, Horváth B, Zsengellėr Z, Bátkai S, Cao Z, Kechrid M, et al. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic Biol Med 2012;53:1123–38.ArticlePubMedPMC
- 43. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279–89.ArticlePubMedPMC
- 44. Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 1983;67:1016–23.ArticlePubMed
- 45. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005;201:1135–43.ArticlePubMedPMC
- 46. Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol 2004;173:7115–9.ArticlePubMed
- 47. Karwinski W, Søreide O. Allopurinol improves scavenging ability of the liver after ischemia/reperfusion injury. Liver 1997;17:139–43.ArticlePubMed
- 48. Liu PG, He SQ, Zhang YH, Wu J. Protective effects of apocynin and allopurinol on ischemia/reperfusion-induced liver injury in mice. World J Gastroenterol 2008;14:2832–7.ArticlePubMedPMC
- 49. Abe T, Unno M, Takeuchi H, Kakita T, Katayose Y, Rikiyama T, et al. A new free radical scavenger, edaravone, ameliorates oxidative liver damage due to ischemia-reperfusion in vitro and in vivo. J Gastrointest Surg 2004;8:604–15.ArticlePubMed
- 50. He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002;512:1–7.ArticlePubMed
- 51. Bilzer M, Lauterburg BH. Effects of hypochlorous acid and chloramines on vascular resistance, cell integrity, and biliary glutathione disulfide in the perfused rat liver: modulation by glutathione. J Hepatol 1991;13:84–9.ArticlePubMed
- 52. Atalla SL, Toledo-Pereyra LH, MacKenzie GH, Cederna JP. Influence of oxygen-derived free radical scavengers on ischemic livers. Transplantation 1985;40:584–90.ArticlePubMed
- 53. Nordström G, Säljö A, Hasselgren PO. Studies on the possible role of oxygen-derived free radicals for impairment of protein and energy metabolism in liver ischemia. Circ Shock 1988;26:115–26.PubMed
- 54. Marubayashi S, Dohi K, Yamada K, Kawasaki T. Changes in the levels of endogenous coenzyme Q homologs, alpha-tocopherol, and glutathione in rat liver after hepatic ischemia and reperfusion, and the effect of pretreatment with coenzyme Q10. Biochim Biophys Acta 1984;797:1–9.PubMed
- 55. Betteridge DJ. What is oxidative stress? Metabolism 2000;49(2 Suppl 1):3–8.Article
- 56. Layton ME, Wood JG, Yan ZY, Forster J. Ischemia/reperfusion alters uric acid and ascorbic acid levels in liver. J Surg Res 1996;64:1–5.ArticlePubMed
- 57. Birlouez-Aragon I, Tessier FJ. Antioxidant vitamins and degenerative pathologies. A review of vitamin C. J Nutr Health Aging 2003;7:103–9.PubMed
- 58. Traber MG. Determinants of plasma vitamin E concentrations. Free Radic Biol Med 1994;16:229–39.ArticlePubMed
- 59. Vertuani S, Angusti A, Manfredini S. The antioxidants and pro-antioxidants network: an overview. Curr Pharm Des 2004;10:1677–94.ArticlePubMed
- 60. Mallick IH, Yang W, Winslet MC, Seifalian AM. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci 2004;49:1359–77.ArticlePubMed
- 61. Carini R, Albano E. Recent insights on the mechanisms of liver preconditioning. Gastroenterology 2003;125:1480–91.ArticlePubMed
- 62. Anttila V, Haapanen H, Yannopoulos F, Herajärvi J, Anttila T, Juvonen T. Review of remote ischemic preconditioning: from laboratory studies to clinical trials. Scand Cardiovasc J 2016;50:355–61.ArticlePubMed
- 63. Ocuin LM, Zeng S, Cavnar MJ, Sorenson EC, Bamboat ZM, Greer JB, et al. Nilotinib protects the murine liver from ischemia/reperfusion injury. J Hepatol 2012;57:766–73.ArticlePubMedPMC
- 64. Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci U S A 2009;106:10764–9.ArticlePubMedPMC
- 65. Shiratori Y, Kiriyama H, Fukushi Y, Nagura T, Takada H, Hai K, et al. Modulation of ischemia-reperfusion-induced hepatic injury by Kupffer cells. Dig Dis Sci 1994;39:1265–72.ArticlePubMed
- 66. Panés J, Perry M, Granger DN. Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention. Br J Pharmacol 1999;126:537–50.ArticlePubMedPMC
- 67. Caldwell-Kenkel JC, Currin RT, Tanaka Y, Thurman RG, Lemasters JJ. Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology 1991;13:83–95.ArticlePubMed
- 68. Oliveira TH, Marques PE, Proost P, Teixeira MM. Neutrophils: a cornerstone of liver ischemia and reperfusion injury. Lab Invest 2018;98:51–62.ArticlePubMed
- 69. Yang YL, Li JP, Xu XP, Dou KF, Yue SQ, Li KZ. Protective effects of tumor necrosis factor alpha antibody and ulinastatin on liver ischemic reperfusion in rats. World J Gastroenterol 2004;10:3161–4.ArticlePubMedPMC
- 70. Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am J Physiol 1993;264(4 Pt 1):G801–9.ArticlePubMed
References
Figure & Data
References
Citations
- Thymoquinone-loaded self-nano-emulsifying drug delivery system against ischemia/reperfusion injury
Badr Bahloul, Roua Chaabani, Yosri Zahra, Nesrine Kalboussi, Jamil Kraiem, Souad Sfar, Nathalie Mignet, Hassen ben Abdennebi
Drug Delivery and Translational Research.2024; 14(1): 223. CrossRef - Modafinil lightens apoptosis and inflammatory response in hepatic ischemia‐reperfusion injury through inactivation of TLR9/Myd88/p38 signaling
Tairan Zhang, Xidong Wang
Drug Development Research.2024;[Epub] CrossRef - Cellular and molecular mechanisms of hepatic ischemia-reperfusion injury: the role of oxidative stress and therapeutic approaches
Joseph George, Yongke Lu, Mutsumi Tsuchishima, Mikihiro Tsutsumi
Redox Biology.2024; : 103258. CrossRef - An update on the molecular mechanism and pharmacological interventions for Ischemia-reperfusion injury by regulating AMPK/mTOR signaling pathway in autophagy
Bin Tang, Zhijian Luo, Rong Zhang, Dongmei Zhang, Guojun Nie, Mingxing Li, Yan Dai
Cellular Signalling.2023; : 110665. CrossRef - Unveiling the Crucial Roles of O2•–and ATP in Hepatic Ischemia–Reperfusion Injury Using Dual-Color/Reversible Fluorescence Imaging
Jihong Liu, Wen Zhang, Xin Wang, Qi Ding, Chuanchen Wu, Wei Zhang, Luling Wu, Tony D. James, Ping Li, Bo Tang
Journal of the American Chemical Society.2023; 145(36): 19662. CrossRef - New insights into ischemia-reperfusion injury signaling pathways in organ transplantation
Kenneth J. Dery, Jerzy W. Kupiec-Weglinski
Current Opinion in Organ Transplantation.2022; 27(5): 424. CrossRef - Isolongifolene alleviates liver ischemia/reperfusion injury by regulating AMPK-PGC1α signaling pathway-mediated inflammation, apoptosis, and oxidative stress
Jinjin Li, Jie Li, Hongbo Fang, Hang Yang, Tianchun Wu, Xiaoyi Shi, Chun Pang
International Immunopharmacology.2022; 113: 109185. CrossRef - Molecularly Designed Ion-Imprinted Nanoparticles for Real-Time Sensing of Cu(II) Ions Using Quartz Crystal Microbalance
Nihan Aydoğan, Gülgün Aylaz, Monireh Bakhshpour, Tugba Tugsuz, Müge Andaç
Biomimetics.2022; 7(4): 191. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite