E-Submission
E-SubmissionIndexed in: ESCI, Scopus, PubMed,
PubMed Central, CAS, DOAJ, KCI
PubMed Central, CAS, DOAJ, KCI
FREE article processing charge

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 37(2); 2020 > Article
-
Review article
Function of hepatocyte growth factor in gastric cancer proliferation and invasion -
Sung Ae Koh
 , Kyung Hee Lee
, Kyung Hee Lee -
Yeungnam University Journal of Medicine 2020;37(2):73-78.
DOI: https://doi.org/10.12701/yujm.2019.00437
Published online: February 20, 2020
Department of Hematology-Oncology, Yeungnam University College of Medicine, Daegu, Korea
- Corresponding author: Kyung Hee Lee Department of Hematology-Oncology, Yeungnam University College of Medicine, 170 Hyeonchung-ro, Nam-gu, Daegu 42415, Korea Tel: +82-53-620-3845 Fax: +82-53-654-8386 E-mail: lkhee@med.yu.ac.kr
• Received: December 13, 2019 • Revised: January 21, 2020 • Accepted: January 28, 2020
Copyright © 2020 Yeungnam University College of Medicine
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 6,053 Views
- 150 Download
- 6 Crossref
Abstract
- Cancer incidence has been increasing steadily and is the leading cause of mortality worldwide. Gastric cancer is still most common malignancy in Korea. Cancer initiation and progression are multistep processes involving various growth factors and their ligands. Among these growth factors, we have studied hepatocyte growth factor (HGF), which is associated with cell proliferation and invasion, leading to cancer and metastasis, especially in gastric cancer. We explored the intercellular communication between HGF and other surface membrane receptors in gastric cancer cell lines. Using complimentary deoxyribonucleic acid microarray technology, we found new genes associated with HGF in the stomach cancer cell lines, NUGC-3 and MKN-28, and identified their function within the HGF pathway. The HGF/N-methyl-N’-nitroso-guanidine human osteosarcoma transforming gene (c-MET) axis interacts with several molecules including E-cadherin, urokinase plasminogen activator, KiSS-1, Jun B, and lipocalin-2. This pathway may affect cell invasion and metastasis or cell apoptosis and is therefore associated with tumorigenesis and metastasis in gastric cancer.
- Cancer initiation and progression involve a multistep process. Cancer initiation requires germline mutation, amplified oncogene, mutated suppressor genes, or hormone action, while cancer progression and metastasis require various growth factors, including epidermal growth factor (EGF), hepatocyte growth factor (HGF), and vascular endothelial growth factors (VEGFs), as well as proteases and adhesion molecules (Table 1) [1-6]. Cell surface receptors can bind growth factors and other ligands, which activate the receptors and transduce the signals by activating a tyrosine kinase inhibitor, thereby regulating cell functions such as cell survival, cell proliferation, protein synthesis, and angiogenesis [7].
- HGF, an effector on cells expressing the N-methyl-N’-nitroso-guanidine human osteosarcoma transforming gene (c-MET) tyrosine kinase receptor, is produced by mesenchymal cells and acts on cells of epithelial origin in paracrine or autonomic fashion [8]. Studies have shown that overexpression or over-activation of HGF can lead to misplaced or inappropriately timed angiogenic and mitogenic signals. c-MET is a cell surface membrane receptor composed of a 50 kDa α-chain and a 145 kDa β-chain [9]. MET activity is observed during embryogenesis and organogenesis in normal cells and is also activated in degenerative diseases such lung and renal fibrosis and liver cirrhosis [10]. Although the HGF/c-MET axis plays a principal role in normal cell development, aberrant activation of this axis is thought to be involved in cell invasion and metastasis in most types of human cancers [11]. We have studied the HGF/c-MET pathway and the associated tumor invasion and proliferation in gastric cancer for several years and here, we review our experiment results.
Introduction
- 1. E-cadherin and β-catenin
- MET has been demonstrated to interact with other cell surface receptors, including integrins, human epidermal growth factor receptor, and FAS receptor, to enhance downstream signaling and tumorigenesis. We previously reported that the expression of E-cadherin (ECD) and urokinase plasminogen activator (uPA) is associated with the development of pancreatic cancer [12]. ECD is a transmembrane glycoprotein that is responsible for calcium-dependent intercellular adhesion by homotypic interaction and is one of the principal elements of the cytoskeleton. Decrease or loss of ECD is frequently associated with cell-to-cell disengagement, tumor invasion, and metastasis [13,14]. ECD functions to dephosphorylate β-catenin, thus inhibiting the binding of intracellular ECD to catenin proteins. It has been suggested that HGF reduces cell-to-cell adhesion by dephosphorylation of the ECD/catenin complex and ECD shedding [15]. We hypothesized that HGF/c-MET may interact with ECD to promote tumorigenesis through the activation of matrix metalloproteinase-7 (MMP-7), which degrades many cellular matrix proteins and adhesion molecules (Fig. 1). To confirm this hypothesis, we investigated the association between HGF/c-MET, ECD, and MMP-7 in two stomach cancer cell lines, NUGC-3 and MKN-28. Western blot and reverse transcription PCR analyses showed that treatment of these cells with HGF reduced the expression of ECD. These results suggested that HGF may stimulate the extracellular cleavage of ECD, thereby increasing the shedding of the soluble fragment and decreasing the 120-kDa full-length ECD in the total cell lysates [16]. MMP-7, which is known to be expressed predominantly by tumor cells in various cancers, was increased by HGF treatment and knockdown of MMP-7 expression in the stomach cancer cell lines resulted in no extracellular cleavage of ECD as well as decreased in vitro cell invasion. These results suggest that HGF may interact with ECD, leading to the activation of the MMP-7 pathway and increased cell invasion.
- 2. Urokinase plasminogen activator
- uPA is a member of the family of serine proteases and is known to participate in cell migration and tissue remodeling. uPA overexpression has been reported in lung, colon, and breast cancers [17-19]. Many studies have shown that blocking the expression of uPA or inhibiting its binding to the uPA receptor (uPAR) suppresses tumor cell invasion and metastasis in various cancer cell lines [20,21]. We measured uPAR expression in 26 patients with stomach cancer before and after surgery and found that uPAR expression was significantly decreased after surgery (p<0.05). We also found that the survival rate of patients with gastric tumors expressing uPAR was significantly lower than that of patients with tumors not expressing uPAR (p=0.035) [22]. We hypothesized that uPA is also associated with tumor progression in gastric cancer and we explored the relationship between HGF and uPA in gastric cancer tumorigenesis. We found that HGF induced reactive oxygen species generation, which regulates uPA production and tumor invasion via mitogen-activated protein (MAP) kinase [23]. Previous studies have also examined the connection between HGF and uPA. One study showed that histone deacetylase (HDAC) regulates HGF-induced expression of both uPA and MMP-9 through a protein kinase C (PKC) dependent pathway in gastric cancer [24]. Another study showed that survivin, a member of the inhibitor of apoptosis family, increases HGF-induced uPA expression and seems to play a role in gastric cancer tumorigenesis [25].
- 3. New HGF regulatory genes
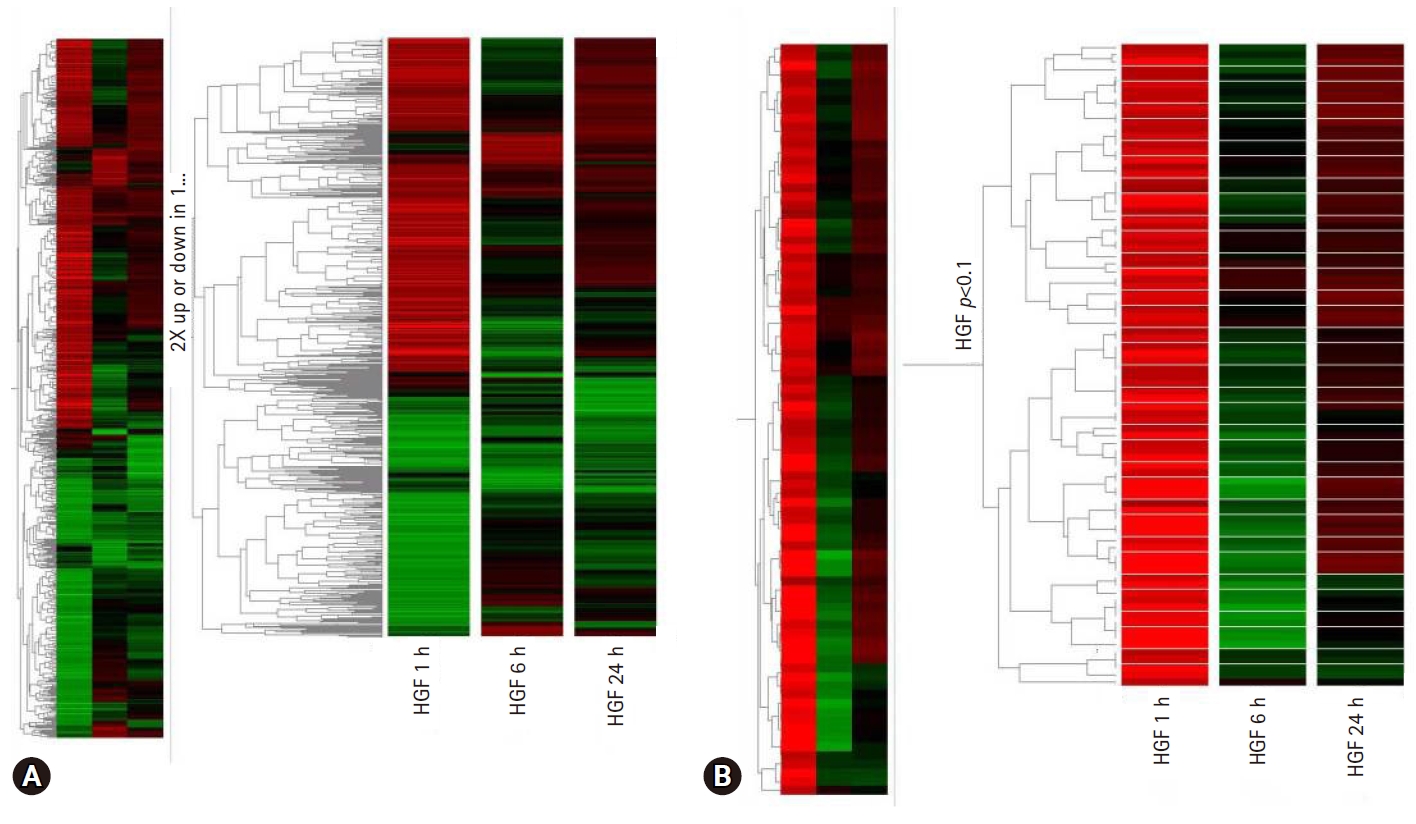
- To find new HGF regulatory genes and identify their role in HGF-induced stomach cancer cell survival, we screened for genes induced by HGF using complimentary deoxyribonucleic acid (cDNA) microarray technology in the stomach cancer cell lines, NUGC-3 and MKN-28 (Fig. 2). We selected the genes that were up or downregulated by more than three-fold in NUGC-3 and MKN-28 cells during HGF treatment (Table 2) and determined their function in conjunction with HGF.
- Bcl-2 associated agonist of cell death (BAD), a BH3-only proapoptotic Bcl-2 family protein, has been found to be upregulated in response to HGF treatment. BAD functions by inactivating anti-apoptotic Bcl-2 proteins [26]. cDNA microarray analysis results have confirmed that BAD is upregulated at the RNA and protein levels following HGF treatment. Our data showed that HGF induced BAD overexpression and enhanced BAD phosphorylation, thereby inhibiting apoptosis and promoting cancer cell survival [27].
- KiSS-1 was also upregulated in response to HGF treatment. KiSS-1 is a putative metastasis suppressor gene and its expression is increased in several human malignancies including melanoma [28] and breast cancer [29]. One study also reported that overexpression of KiSS-1 in breast cancer cells results in a more aggressive phenotype [30]. Consistent with these results, we found that HGF induced the overexpression of KiSS-1 in a p38-dependent manner. In addition, KiSS-1 suppressed MMP-9 expression and decreased cell invasion in vitro, suggesting it may act as a metastasis suppressor gene in gastric cancer [31].
- Jun B was also upregulated in response to HGF treatment. Jun B belongs to the June gene family (c-Jun, JunB, and JunD), whose members encode the activator protein-1 (AP-1) family of transcription factors. AP-1 is a dimeric transcription factor that is enhanced by the MAP kinase pathway in the presence of growth factors, hormones, or other environmental stresses [32,33]. Of the AP-1 components, c-Jun and c-Fos were first identified as viral oncoproteins; thus, their function in tumorigenesis has been established. However, it is also known that some Jun and Fos proteins can suppress tumor formation [34]. Accordingly, we examined the role of Jun B in gastric cancer. In our study, Jun B levels were decreased by inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and cell proliferation and invasion were decreased in Jun B knockdown stomach cancer cell lines. Further, Jun B knockdown cells blocked the MMP-9 upregulation induced by HGF. MMP-9 is a matrix metalloproteinase protein that degrades the basement membrane, exposing cryptic sites within the matrix and resulting in cancer cell invasion [35,36]. These data suggest that Jun B expression induced by HGF can activate MMP-9 by the NF-κB pathway and thereby contribute to invasion and cell proliferation in gastric cancer [37].
- We recently studied that lipocalin-2 (LCN2) is upregulated by HGF treatment. LCN2 is a member of the lipocalin family, which binds and transports small lipophilic molecules including leukotrienes, retinoic acids, and prostaglandins, and it was first identified as a modulator of the immune system [38]. In addition, LCN2 binds MMP-9, forming a complex comprising LCN2 and MMP-9, promoting MMP-9 activation, and preventing its degradation [35,36]. HGF treatment upregulated the expression of LCN2 in gastric cancer cells, leading to increased activation of MMP-9. Knockdown of LCN2 in these cells decreased MMP-9 activation in response to HGF treatment and treatment of the cells with an NF-κB inhibitor prevented the HGF-mediated upregulation in LCN2 expression. Further, HGF-mediated cell proliferation and invasion was decreased in LCN2 knockdown cells compared to control cells [39]. These data suggest that HGF induces the upregulation of LCN2 expression, which activates MMP-9, and HGF may play a role in proliferation and invasion of gastric cancer.
Interaction of HGF and other molecular proteins in gastric cancer
- Abberant activation of MET signaling occurs in a subset of advanced cancers, including gastric cancer. The HGF/c-MET axis interacts with several molecules including ECD, uPA, KiSS-9, Jun B, and LCN2. This pathway may affect cell invasion and metastasis or cell apoptosis and is therefore associated with tumorigenesis and metastasis in gastric cancer, which maybe one of the important therapeutic targets. To validate our findings, further experiments are warranted using in vivo knockout mouse models.
Conclusion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Author contributions
Conceptualization: SAK, KHL; Data curation: KHL; Formal analysis: SAK; Project administration: KHL; Validation: SAK; Writing–original draft: SAK; Writing–review & editing: SAK, KHL.
Notes
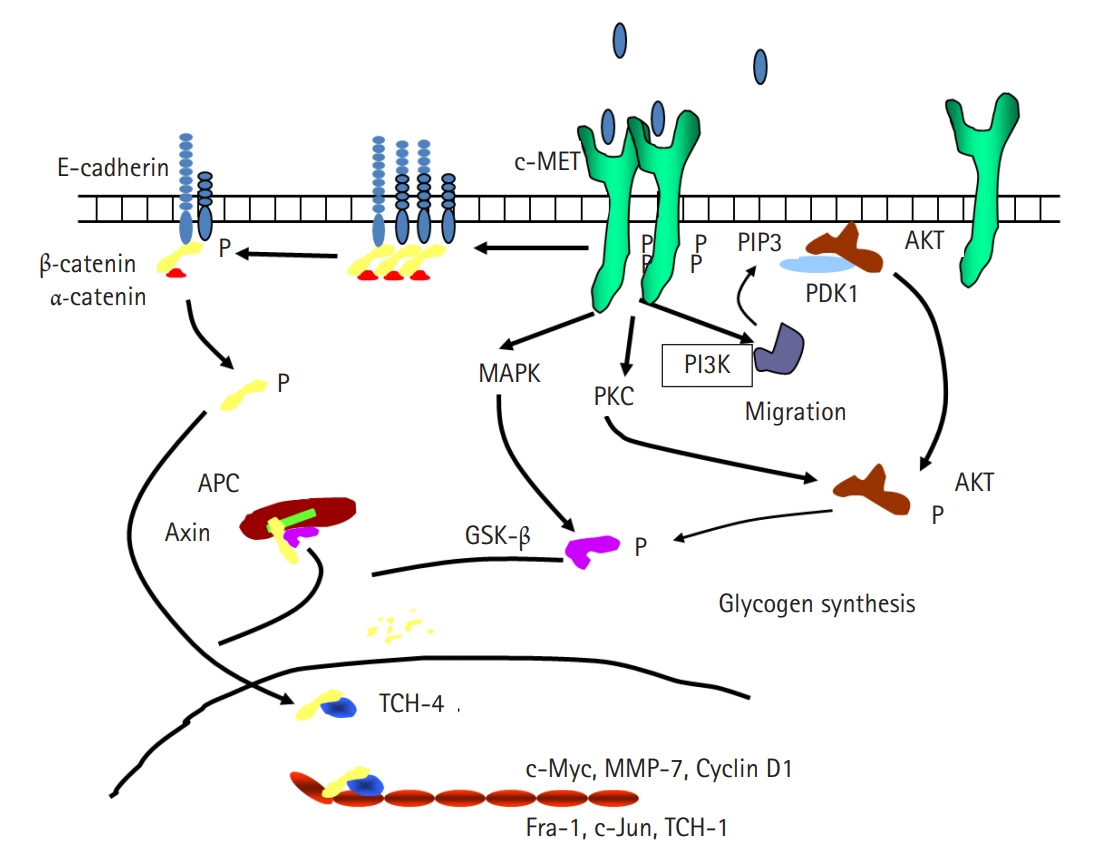
Fig. 1.Schematic diagram of the relationship between hepatocyte growth factor and E-cadherin. APC, adenomatous synthase kinase-binding protein; AKT, protein kinase B; c-MET, N-methyl-N’-nitroso-guanidine human osteosarcoma transforming gene tyrosin kinase receptor; GSK-β, glycogen synthase kinase β; MAPK, mitogen-activated protein kinase; MMP-7, matrix metalloproteinase-7; PDK1, phosphoinositide-dependent kinase-1; PI3K, phosphinositol-3 kinase; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; TCF-1, transcription factor 1; TCF-4, transcription factor 4.
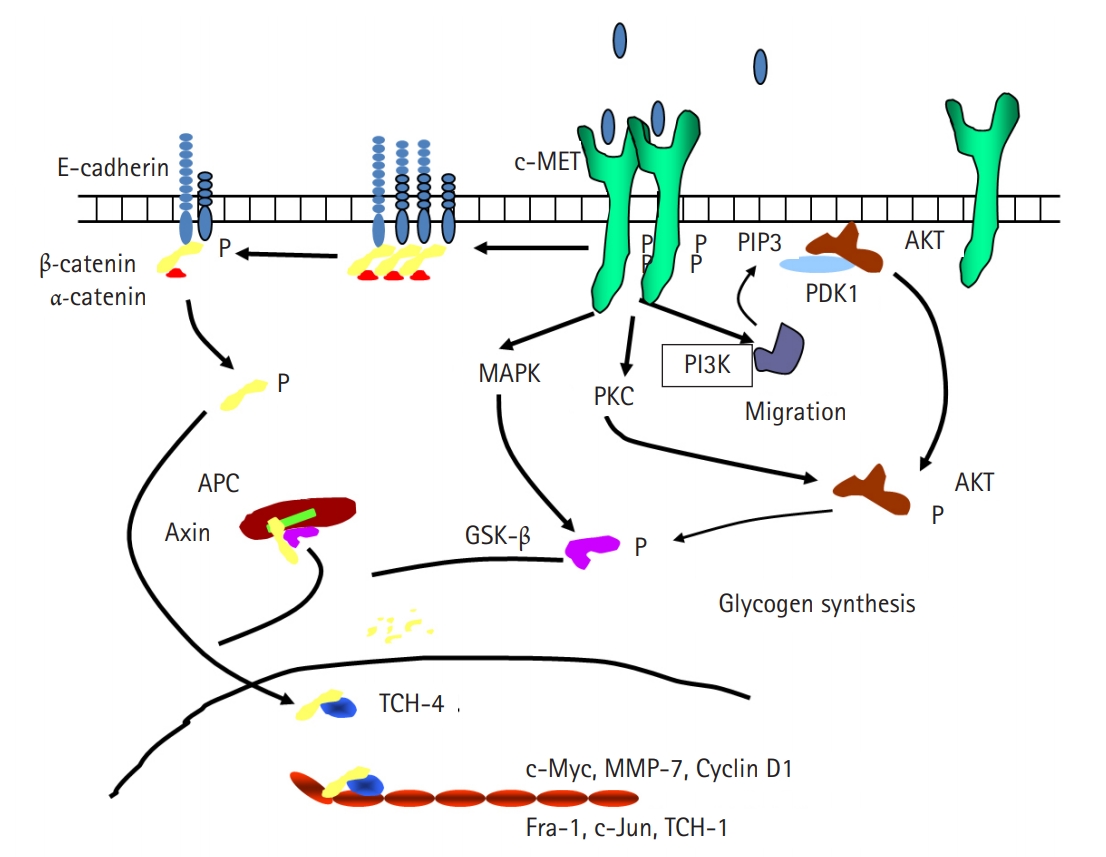
Fig. 2.(A) Genetree showing genes up or downregulated by at least 2-fold after 1 hour, 6 hours, and 24 hours of hepatocyte growth factor (HGF) treatment. (B) Genetree of t-test.
Table 1.Gowth factors in cancer progression
| Growth factor | Property | Receptor | Study |
|---|---|---|---|
| Vascular endothelial growth factor | Endothelial mitogen, survival factor, and permeability inducer produced by many types of tumor cells | Flk-1/KDR (VEGFR-2), Flt-1 (VEGFR-1) (both present on activated endothelium) | Veikkola and Alitalo [1] |
| Transforming growth factor-α (TGF-α) | Endothelial mitogen and angiogenesis inducer; inducer of vascular endothelial growth factor expression | Epidermal growth factor-R | Schmitt and Soares [2] |
| Fibroblast growth factor | Endothelial mitogen, angiogenesis inducer, and survival factor; inducer of Flk-1 expression | FGF-RI-4 | Botta et al. [3] |
| Epidermal growth factor (EGF) | Weak endothelial mitogen; inducer of vascular endothelial growth factor expression | Epidermal growth factor-R | Mooradian and Diglio [4] |
| Hepatocyte growth factor/scatter factor (HGF/SF) | Endothelial mitogen, motogen, and angiogenesis inducer | c-MET | Lamszus et al. [5] |
| Interleukin-8 | In vivo-acting, possibly indirect angiogenesis inducer | Interleukin-8R presence on endothelial cells remains uncertain | Desbaillets et al. [6] |
Table 2.Genes induced by hepatocyte growth factor using complimentary deoxyribonucleic acid microarray
- 1. Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol 1999;9:211–20.ArticlePubMed
- 2. Schmitt FC, Soares R. TGF-alpha and angiogenesis. Am J Surg Pathol 1999;23:358–9.ArticlePubMed
- 3. Botta M, Manetti F, Corelli F. Fibroblast growth factors and their inhibitors. Curr Pharm Des 2000;6:1897–924.ArticlePubMed
- 4. Mooradian DL, Diglio CA. Production of a transforming growth factor-beta-like growth factor by RSV-transformed rat cerebral microvascular endothelial cells. Tumour Biol 1991;12:171–83.ArticlePubMed
- 5. Lamszus K, Jin L, Fuchs A, Shi E, Chowdhury S, Yao Y, et al. Scatter factor stimulates tumor growth and tumor angiogenesis in human breast cancers in the mammary fat pads of nude mice. Lab Invest 1997;76:339–53.PubMed
- 6. Desbaillets I, Diserens AC, Tribolet N, Hamou MF, Van Meir EG. Upregulation of interleukin 8 by oxygen-deprived cells in glioblastoma suggests a role in leukocyte activation, chemotaxis, and angiogenesis. J Exp Med 1997;186:1201–12.ArticlePubMedPMCPDF
- 7. Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 1999;82:241–50.ArticlePubMed
- 8. Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995;373:699–702.ArticlePubMedPDF
- 9. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010;11:834–48.ArticlePubMedPDF
- 10. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012;12:89–103.ArticlePubMedPDF
- 11. Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 2002;13:41–59.ArticlePubMed
- 12. Shin SJ, Kim KO, Kim MK, Lee KH, Hyun MS, Kim KJ, et al. Expression of E-cadherin and uPA and their association with the prognosis of pancreatic cancer. Jpn J Clin Oncol 2005;35:342–8.ArticlePubMedPDF
- 13. Chen WC, Obrink B. Cell-cell contacts mediated by E-cadherin (uvomorulin) restrict invasive behavior of L-cells. J Cell Biol 1991;114:319–27.ArticlePubMedPMCPDF
- 14. Althaus E, Karotke E, Nitsch K, Winkler H. An experimental re-examination of the upper stability limit of muscovite plus quartz. Neues Jahrb Miner Monatsh 1970;7:325–36.
- 15. Hiscox S, Jiang WG. Hepatocyte growth factor/scatter factor disrupts epithelial tumour cell-cell adhesion: involvement of beta-catenin. Anticancer Res 1999;19:509–17.PubMed
- 16. Lee KH, Choi EY, Hyun MS, Jang BI, Kim TN, Kim SW, et al. Association of extracellular cleavage of E-cadherin mediated by MMP-7 with HGF-induced in vitro invasion in human stomach cancer cells. Eur Surg Res 2007;39:208–15.ArticlePubMed
- 17. Markus G, Takita H, Camiolo SM, Corasanti JG, Evers JL, Hobika GH. Content and characterization of plasminogen activators in human lung tumors and normal lung tissue. Cancer Res 1980;40:841–8.PubMed
- 18. Sappino AP, Busso N, Belin D, Vassalli JD. Increase of urokinase-type plasminogen activator gene expression in human lung and breast carcinomas. Cancer Res 1987;47:4043–6.PubMed
- 19. Markus G, Camiolo SM, Kohga S, Madeja JM, Mittelman A. Plasminogen activator secretion of human tumors in short-term organ culture, including a comparison of primary and metastatic colon tumors. Cancer Res 1983;43:5517–25.PubMed
- 20. Alonso DF, Farias EF, Ladeda V, Davel L, Puricelli L, Bal de Kier Joffe E. Effects of synthetic urokinase inhibitors on local invasion and metastasis in a murine mammary tumor model. Breast Cancer Res Treat 1996;40:209–23.ArticlePubMedPDF
- 21. Kruger A, Soeltl R, Lutz V, Wilhelm OG, Magdolen V, Rojo EE, et al. Reduction of breast carcinoma tumor growth and lung colonization by overexpression of the soluble urokinase-type plasminogen activator receptor (CD87). Cancer Gene Ther 2000;7:292–9.ArticlePubMedPDF
- 22. Lee KH, Bae SH, Lee JL, Hyun MS, Kim SH, Song SK, et al. Relationship between urokinase-type plasminogen receptor, interleukin-8 gene expression and clinicopathological features in gastric cancer. Oncology 2004;66:210–7.ArticlePubMed
- 23. Lee KH, Kim SW, Kim JR. Reactive oxygen species regulate urokinase plasminogen activator expression and cell invasion via mitogen-activated protein kinase pathways after treatment with hepatocyte growth factor in stomach cancer cells. J Exp Clin Cancer Res 2009;28:73.ArticlePubMedPMC
- 24. Lee KH, Choi EY, Kim MK, Kim KO, Jang BI, Kim SW, et al. Inhibition of histone deacetylase activity down-regulates urokinase plasminogen activator and matrix metalloproteinase-9 expression in gastric cancer. Mol Cell Biochem 2010;343:163–71.ArticlePubMedPDF
- 25. Lee KH, Choi EY, Koh SA, Kim MK, Kim KO, Lee SH, et al. Down-regulation of survivin suppresses uro-plasminogen activator through transcription factor JunB. Exp Mol Med 2011;43:501–9.ArticlePubMedPMC
- 26. Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995;80:285–91.ArticlePubMed
- 27. Lee KH, Choi EY, Kim MK, Hyun MS, Eun JR, Jang BI, et al. Hepatocyte growth factor promotes cell survival by phosphorylation of BAD in gastric cancer cells. Oncol Res 2008;17:23–32.ArticlePubMed
- 28. Lee JH, Welch DR. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int J Cancer 1997;71:1035–44.ArticlePubMed
- 29. Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res 1997;57:2384–7.PubMed
- 30. Martin TA, Watkins G, Jiang WG. KiSS-1 expression in human breast cancer. Clin Exp Metastasis 2005;22:503–11.ArticlePubMedPDF
- 31. Lee KH, Kim JR. Kiss-1 suppresses MMP-9 expression by activating p38 MAP kinase in human stomach cancer. Oncol Res 2009;18:107–16.ArticlePubMed
- 32. Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Transcription factors control invasion: AP-1 the first among equals. Oncogene 2007;26:1–10.ArticlePubMedPDF
- 33. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003;3:859–68.ArticlePubMedPDF
- 34. Vandel L, Pfarr CM, Huguier S, Loiseau L, Sergeant A, Castellazzi M. Increased transforming activity of JunB and JunD by introduction of an heterologous homodimerization domain. Oncogene 1995;10:495–507.PubMed
- 35. Tschesche H, Zolzer V, Triebel S, Bartsch S. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. Eur J Biochem 2001;268:1918–28.ArticlePubMed
- 36. Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem 2001;276:37258–65.ArticlePubMed
- 37. Lee KH, Kim JR. Regulation of HGF-mediated cell proliferation and invasion through NF-κB, JunB, and MMP-9 cascades in stomach cancer cells. Clin Exp Metastasis 2012;29:263–72.ArticlePubMedPDF
- 38. Bratt T, Ohlson S, Borregaard N. Interactions between neutrophil gelatinase-associated lipocalin and natural lipophilic ligands. Biochim Biophys Acta 1999;1472:262–9.ArticlePubMed
- 39. Koh SA, Lee KH. HGF mediated upregulation of lipocalin 2 regulates MMP9 through nuclear factor-κB activation. Oncol Rep 2015;34:2179–87.ArticlePubMed
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Reciprocal Regulation of Cancer-Associated Fibroblasts and Tumor Microenvironment in Gastrointestinal Cancer: Implications for Cancer Dormancy
Shih-Hsuan Cheng, Hsin-Ying Clair Chiou, Jiunn-Wei Wang, Ming-Hong Lin
Cancers.2023; 15(9): 2513. CrossRef - Expression of E-cadherin and N-cadherin in Epithelial-to-Mesenchymal Transition of Osteosarcoma: A Systematic Review
Leo Issagholian, Ethan Tabaie, Akshay J Reddy, Muhammad S Ghauri, Rakesh Patel
Cureus.2023;[Epub] CrossRef - Remodelling of the tumour microenvironment by the kallikrein-related peptidases
Srilakshmi Srinivasan, Thomas Kryza, Jyotsna Batra, Judith Clements
Nature Reviews Cancer.2022; 22(4): 223. CrossRef - Identification of hub pathways and drug candidates in gastric cancer through systems biology
Seyed Reza Salarikia, Mohammad Kashkooli, Mohammad Javad Taghipour, Mahdi Malekpour, Manica Negahdaripour
Scientific Reports.2022;[Epub] CrossRef - Signaling pathways and therapeutic interventions in gastric cancer
Zi-Ning Lei, Qiu-Xu Teng, Qin Tian, Wei Chen, Yuhao Xie, Kaiming Wu, Qianlin Zeng, Leli Zeng, Yihang Pan, Zhe-Sheng Chen, Yulong He
Signal Transduction and Targeted Therapy.2022;[Epub] CrossRef - Proteomic Signatures of Diffuse and Intestinal Subtypes of Gastric Cancer
Smrita Singh, Mohd Younis Bhat, Gajanan Sathe, Champaka Gopal, Jyoti Sharma, Anil K. Madugundu, Neha S. Joshi, Akhilesh Pandey
Cancers.2021; 13(23): 5930. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite