E-Submission
E-SubmissionPubMed Central, CAS, DOAJ, KCI

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 41(2); 2024 > Article
-
Review article
Comprehensive overview of the role of mitochondrial dysfunction in the pathogenesis of acute kidney ischemia-reperfusion injury: a narrative review -
Min-Ji Kim1,*
 , Chang Joo Oh2,*, Chang-Won Hong3, Jae-Han Jeon1
, Chang Joo Oh2,*, Chang-Won Hong3, Jae-Han Jeon1 -
Journal of Yeungnam Medical Science 2024;41(2):61-73.
DOI: https://doi.org/10.12701/jyms.2023.01347
Published online: February 14, 2024
1Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Korea
2Research Institute of Aging and Metabolism, School of Medicine, Kyungpook National University, Daegu, Korea
3Department of Physiology, School of Medicine, Kyungpook National University, Daegu, Korea
- Corresponding author: Jae-Han Jeon, MD, PhD Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, 807 Hoguk-ro, Buk-gu, Daegu 41404, Korea Tel: +82-53-200-7201 • Fax: +82-53-200-3155 • E-mail: jeonjh@knu.ac.kr
- *These authors contributed equally to this work as co-first authors.
© 2024 Yeungnam University College of Medicine, Yeungnam University Institute of Medical Science
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,977 Views
- 98 Download
Abstract
- Acute kidney ischemia-reperfusion (IR) injury is a life-threatening condition that predisposes individuals to chronic kidney disease. Since the kidney is one of the most energy-demanding organs in the human body and mitochondria are the powerhouse of cells, mitochondrial dysfunction plays a central role in the pathogenesis of IR-induced acute kidney injury. Mitochondrial dysfunction causes a reduction in adenosine triphosphate production, loss of mitochondrial dynamics (represented by persistent fragmentation), and impaired mitophagy. Furthermore, the pathological accumulation of succinate resulting from fumarate reduction under oxygen deprivation (ischemia) in the reverse flux of the Krebs cycle can eventually lead to a burst of reactive oxygen species driven by reverse electron transfer during the reperfusion phase. Accumulating evidence indicates that improving mitochondrial function, biogenesis, and dynamics, and normalizing metabolic reprogramming within the mitochondria have the potential to preserve kidney function during IR injury and prevent progression to chronic kidney disease. In this review, we summarize recent advances in understanding the detrimental role of metabolic reprogramming and mitochondrial dysfunction in IR injury and explore potential therapeutic strategies for treating kidney IR injury.
- Acute kidney injury (AKI) is a common clinical condition that affects individuals of all ages ranging from children to those who are older [1]. Hospitalized patients, especially those in intensive care units, are at higher risk of developing AKI because of conditions such as surgeries, infections, and medications that can trigger kidney injury. AKI is associated with high morbidity and mortality rates. Severe AKI may progress to end-stage kidney disease or require kidney replacement therapy [2]. Nevertheless, therapies for AKI remain conservative, highlighting the urgent need for innovative therapeutic approaches.
- Kidney ischemia-reperfusion (IR) injury is a pathological condition characterized by an initial reduction in blood supply to the kidneys and subsequent reoxygenation [3]. Counterintuitively, this restoration of blood flow often exacerbates tissue damage, resulting in glomerular and tubular damage, and triggering a severe inflammatory response, termed “reperfusion injury.”
- Mitochondria, the powerhouses of cells, are pivotal for adenosine triphosphate (ATP) generation through oxidative phosphorylation. In normal mitochondrial physiology, the process encompasses not only ATP synthesis via the tricarboxylic acid cycle and redox management but also the handling of reactive oxygen species (ROS). Additionally, processes such as mitochondrial biogenesis and dynamics involving fission, fusion, and distribution are crucial for balancing mitochondrial network maintenance, quality control, and mitochondrial DNA distribution, which are linked to metabolic status [4]. Cellular and organismal health depends on this balance, with significant alterations occurring in response to stress and disease. The mitochondrial death pathway also plays a vital role in maintaining mitochondrial integrity and overall cell health [5].
- Emerging research suggests that mitochondrial dysfunction plays a pivotal role in the pathogenesis of kidney IR injury [6]. The kidneys are high energy-demanding organs because of their roles in filtration, reabsorption, and secretion [7], and they depend heavily on mitochondria. Because mitochondria are primary suppliers of cell energy in the form of ATP, their dysfunction can critically impair the energy supply required for optimal kidney function [6]. Recent studies have suggested that the aberrant ROS levels implicated in tubular and glomerular injuries are associated with mitochondrial dysfunction [8,9].
- In this review, we highlight insights from studies on the interaction between mitochondria and kidney IR injury, elucidating the intricate pathophysiology of the disease and therapeutic strategies. Specifically, we discuss the detrimental roles of mitochondrial dynamics (fusion-fission), mitophagy, and metabolic rewiring, along with the effects of mitochondrial ROS on kidney IR injury.
Introduction
- Kidney IR injury occurs when there is a temporary reduction in blood supply to the kidneys, followed by restoration. It can be triggered by surgical procedures, trauma, or shock. The interruption of blood flow can lead to oxygen and nutrient deprivation, resulting in cellular stress. When the blood flow is restored, a burst of ROS is triggered, which exacerbates oxidative stress and inflammation. Eventually, this sequence of events can damage kidney tubular and endothelial cells, contributing to the development of AKI. Under these conditions, significant pathological changes in the mitochondria occur, manifesting as impaired mitochondrial energetics, excessive mitochondrial fragmentation, defective mitophagy, and ROS bursts within the mitochondria.
- 1. Mitochondrial dysfunction and related cell death
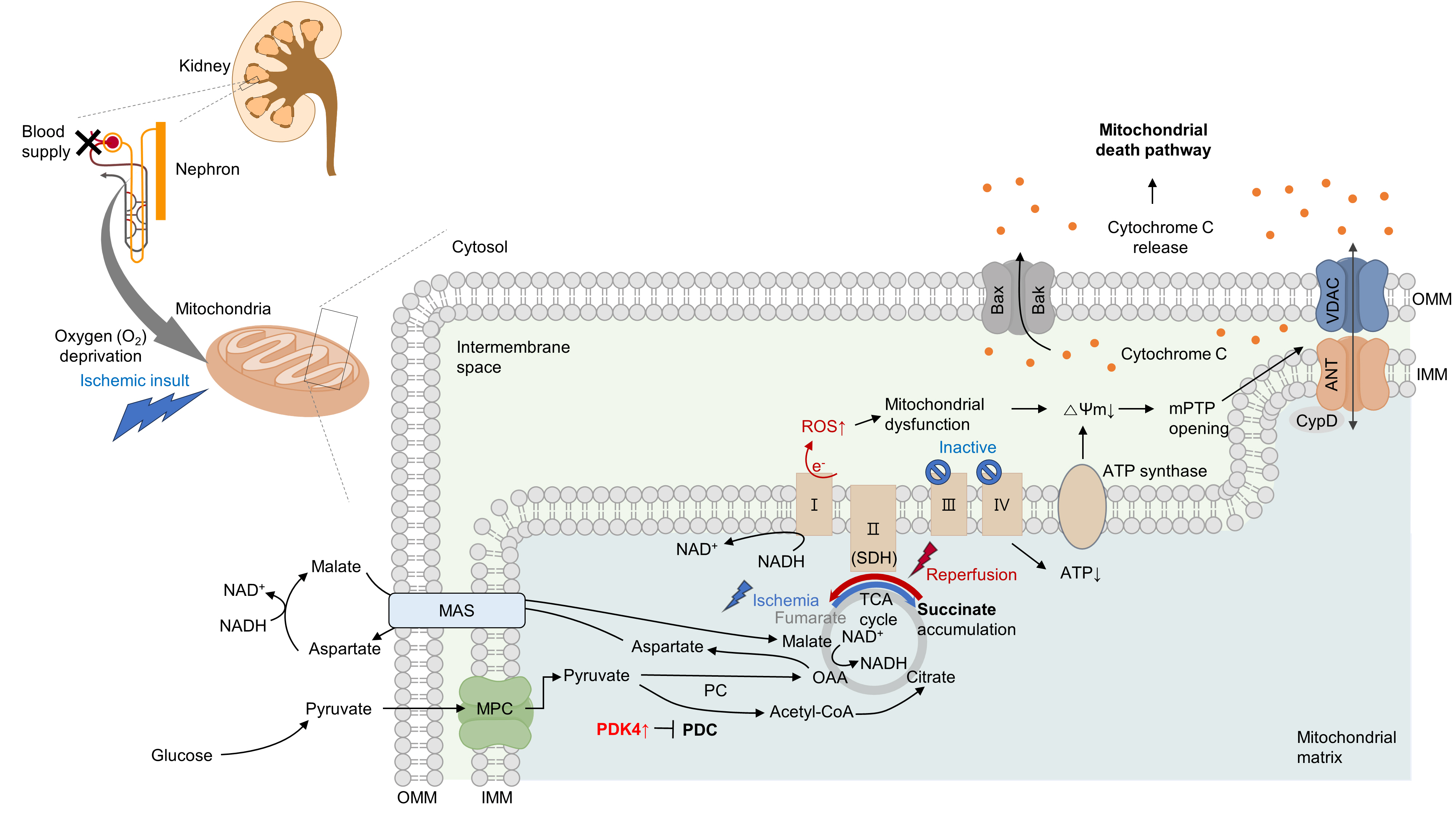
- Several studies have suggested that mitochondrial energetics are compromised following an ischemic insult. This is manifested by a marked reduction in mitochondrial ATP synthesis and mitochondrial membrane potential, and subsequent opening of the mitochondrial permeability transition pore (mPTP) [10] (Fig. 1). Indeed, upon ischemia induction, ATP levels in proximal tubule cells decrease to a nadir within a few minutes [11]. Decreased ATP levels can reduce the activity of Na+/K+-ATPase, leading to the accumulation of intracellular Na+. Subsequently, this accumulation triggers Ca2+ influx and accumulation within the mitochondria. Following reperfusion, excessive increases in ROS and Ca2+ levels occur within the mitochondria, which can induce mitochondrial dysfunction [12,13]. This triggers cytochrome c release through the mPTP, initiating a caspase cascade and activating the mitochondrial death pathway [14,15]. Cyclophilin D plays a key role in the regulation of mPTP. Cyclophilin D knock-out (KO) mice are resistant to IR injury [14]. The endoplasmic reticulum (ER) and mitochondria share close physical and functional connections. Downregulation of X-box binding protein 1 (XBP1), an ER stress response protein, protects the kidneys from IR injury. Mechanistically, 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation 1 (HRD1), a protein downstream of XBP1, serves as an E3-ligase and facilitates the downregulation of nuclear factor erythroid 2-related factor 2 (NRF2) through the ubiquitination-degradation pathway [16]. XBP1 can also upregulate transcription from the NLR family pyrin domain containing 3 (NLRP3) promoter, highlighting the role of the XBP1-NLRP3 axis in regulating ER-mitochondrial crosstalk in kidneys under IR stress [17].
- Under oxygen-deprived conditions, the activity of complex V is impaired, leading to defects in oxidative phosphorylation. In the IR milieu, excessive ROS production is always accompanied by a reduction in ATP production in the mitochondria [18,19]. However, whether mitochondrial dysfunction is a cause or consequence of excessive mitochondrial ROS production remains unclear. In conclusion, therapeutic strategies targeting kidney IR injury should include the prevention of mitochondria-related cell death.
- 2. Metabolic rewiring and aberrant succinate accumulation within the mitochondria
- Previous studies have examined the role of metabolic rewiring in kidney IR injury [20-22]. During both the early and late stages of reperfusion following ischemia, kidney lactate, and pyruvate levels increase, along with an increase in hexokinase activity, indicative of enhanced glycolysis. Additionally, tubules in the process of normal regeneration and those undergoing atrophy show elevated expression of glycolytic enzymes and inhibitory phosphorylation of mitochondrial pyruvate dehydrogenase (PDH) [22]. Similarly, the progression of IR-induced AKI in human kidney proximal tubular cells (HK-2 cells) is characterized by the disruption of amino acid, nucleotide, and tricarboxylic acid cycle metabolism, and notably, a metabolic shift from fatty acid oxidation (FAO) to glycolytic conversion [20]. It could be hypothesized that the induction of a glycolytic phenotype can serve as an adaptive mechanism following tubular injury, as glycolysis can facilitate biomass synthesis crucial for cellular repair and proliferation. However, the continued presence of this phenotype seems to correlate with an elevated level of fibrosis [21].
- Moreover, in proximal tubular cells, hypoxia can induce the reversal of complex II (succinate dehydrogenase, SDH) activity, resulting in excessive succinate accumulation [8,9]. Specifically, the primary source of succinate is the overflow of fumarate from purine nucleotides and partially from the reversal of the malate/aspartate shuttle [8]. Supporting this idea, our group recently discovered that inhibition of the malate/aspartate shuttle by activating PDH flux can attenuate succinate accumulation [9]. In the heart, however, succinate accumulates via the canonical Krebs cycle and not through reverse SDH activity [23]. Although the reason behind this discrepancy remains elusive (e.g., whether it is organ-specific), a reduction in either SDH activity or succinate accumulation is sufficient to mitigate damage to both the kidney and heart [8,9,23]. Consistent with this finding, a recent study revealed that fumarate can accept electrons through net reversal of the SDH complex [24].
- Following reperfusion, the accumulated succinate is rapidly reoxidized by SDH. Aberrant electron accumulation during the ischemia period spills over not only to complexes III, IX, and V but also to complex I during the reperfusion period. This phenomenon, known as reverse electron transport, results in a burst of ROS leading to cell death. Although strategies to reduce tubular damage are still being established, recent findings suggest that pharmacological targeting of SDH by administering dimethyl malonate during reperfusion can ameliorate kidney IR injury by blocking rapid oxidation by SDH [25]. Therefore, the optimal timing for SDH inhibition to prevent kidney IR injury requires further investigation (Fig. 1).
- 3. Excessive mitochondrial fragmentation with defective mitophagy
- Mitochondria undergo fusion or division in response to various stimuli and stresses [26]. Environmental stresses, including hypoxia, can lead to mitochondrial hyperfragmentation. Previous studies have shown that mitochondria in proximal tubular cells undergo excessive fission [9,27]. Mechanistically, the activation of dynamin-related protein 1 (Drp1)-mediated fission and inhibition of Bak-mediated fusion can promote cleavage of the outer membrane [6,28]. In addition, recent findings have indicated that OMA1 zinc metallopeptidase-mediated proteolysis of optic atrophy 1 (OPA1), a key inner membrane fusion protein, contributes to inner membrane cleavage during cellular stress [29]. Tubular cell apoptosis and AKI can be attenuated by the expression of dominant-negative Drp1 or administration of mitochondrial division inhibitor 1 (mdivi-1), a Drp1 inhibitor [6] (Fig. 2A).
- Consistent with these findings, OMA1 deficiency prevented ischemic AKI by inhibiting mitochondrial fragmentation [27]. In addition, deficiency of Numb, a multifunctional adaptor protein, can increase AKI severity by exacerbating mitochondrial fragmentation through phosphorylation of Drp1 at Ser637 [30]. These findings are supported by other studies. Under stress conditions such as IR injury, Bax-interacting factor 1 (Bif-1, a protein implicated in apoptosis and mitophagy) can regulate the mitochondrial inner membrane by interacting with prohibitin 2, which forms complexes in the inner membrane with prohibitin 1. This interaction, in tandem with prohibitin 1, can disrupt prohibitin complexes, leading to the proteolysis and inactivation of OPA1 [31]. A more recent study revealed that mitochondrial fragmentation can be induced and inhibited by uncoupling protein 2 (UCP2) gain-of-function and loss-of-function mutations, respectively, which correlated with kidney function in mice subjected to kidney IR injury [32]. Latent transforming growth factor-beta binding protein 4 (LTBP4) is upregulated in the kidney tissues of patients with AKI. LTBP4 also regulates transforming growth factor beta activity. Knockdown of LTBP4 aggravates injury by accelerating Drp1-dependent mitochondrial division, injury that can be ameliorated by mdivi-1 treatment [33].
- Mitophagy, defined as the clearance of damaged mitochondria from cells to maintain a healthy mitochondrial population, is required to overcome the pathology of kidney IR injury. In particular, the mitochondrial kinase PTEN-induced kinase 1 (PINK1) and the E3-ubiquitin ligase parkin can serve as sensors of mitochondrial quality. They are activated following membrane depolarization [34]. PINK1 accumulates on defective mitochondria, and its subsequent homodimerization on the outer mitochondrial membrane facilitates the recruitment of parkin from the cytosol, mediating the clearance of damaged mitochondria [35,36]. Once recruited to depolarized mitochondria, parkin-dependent ubiquitination and proteasomal degradation of outer membrane proteins ultimately leads to mitophagy [37] (Fig. 2B).
- Recent studies have shown that activation of mitophagy is essential for mitigating IR injury. Mitophagy is induced in kidney proximal tubular cells in both in vitro and in vivo models of ischemic AKI [38], and genetic disruption of mitophagy exacerbates the injury [38,39]. Another study demonstrated that Beclin-1 peptide pretreatment, which induces both autophagy and mitophagy, can protect mice against kidney IR injury [40]. Consistent with this idea, a recent study showed that the genetic deletion of pannexin 1 can prevent kidney tubular cell death, oxidative stress, and mitochondrial damage after IR injury. This protection was attributed to enhanced mitophagy, which modulates the ATP-P2Y-mammalian target of rapamycin signaling pathway [41]. The depletion of STE20-like kinase 1 can ameliorate kidney IR injury via AMP-activated protein kinase (AMPK)-yes-associated protein (YAP)-OPA1-dependent activation of mitophagy [42]. Furthermore, inhibition of acyl-CoA synthetase family member 2 (ACSF2), which is highly expressed in the mitochondria of kidney tubular cells, protects against kidney IR injury by activating mitophagy in proximal tubular cells [43]. Bcl-2 interacting protein 3 (BNIP3), a noncanonical regulator of mitophagy, has also been implicated in kidney IR injury [44]. Taken together, these findings suggest that the regulation of mitophagy is an important therapeutic strategy against kidney IR injury.
- 4. Ferroptosis
- Ferroptosis is a relatively newly discovered form of cell death [45]. It is an iron-dependent cell death process characterized by the accumulation of lipid peroxides. This process is distinct from that of apoptosis and necrosis [45]. Mitochondria in ferroptotic cells display pathological changes in morphology such as shrinkage and loss of cristae [46]. As suggested by its name, ferroptosis can be triggered by various stimuli that lead to iron overload. A pathological increase in the cellular iron content can accelerate the Fenton reaction, resulting in the production of hydroxyl radicals (•OH) and other ROS. Additionally, excess polyunsaturated fatty acids, deprivation of glutathione, and reduced function of mitochondrial glutathione peroxidase 4 (GPX4), an enzyme that decreases lipid peroxides, can induce ferroptosis by increasing lipid ROS (Fig. 3) [46].
- Accumulating evidence has shown that ferroptosis is an important molecular target for the treatment of kidney IR injury. ROS bursts during reperfusion injuries are associated with ferroptosis [47]. Researchers have found that the aryl hydrocarbon receptor (AhR) is activated during reoxygenation, which leads to ROS production, lipid peroxidation, and ferroptotic cell death [47]. The same researchers have also shown that AhR-mediated ferroptosis is detrimental not only during the reperfusion phase but also during the ischemic phase [48]. Tripartite motif-containing protein 21 (TRIM21) is upregulated in the kidney during IR injury and promotes ferroptosis by ubiquitinating and degrading GPX4. TRIM21 inhibition may be a strategy to reduce IR injury [49]. Inhibition of the ER stress inositol-requiring enzyme 1 (IRE1)/ c-Jun N-terminal kinase (JNK) pathway in kidney tubular epithelial cells (TECs) attenuates ferroptosis in AKI. Intriguingly, inhibition of ferroptosis can also attenuate IRE1/JNK signaling, suggesting a feed-forward loop [50]. Moreover, preserving heme oxygenase 1 (HO-1) expression by inhibiting miR-3587 can attenuate ferroptosis and kidney IR injury, suggesting that ferroptosis is a key detrimental pathological mechanism of IR injury [51].
Molecular mechanisms of kidney ischemia-reperfusion injury: mitochondrial perspective
- 1. Targeting mitochondrial function including reactive oxygen species, biogenesis, and related mitochondrial death pathway
- Several studies have suggested that enhancing mitochondrial quantity or function can be beneficial for retarding or preventing IR injury. Approaches such as mitochondrial gene delivery [52], coenzyme Q10 administration [53], and mitochondrial transplantation [54,55] have been shown to be able to attenuate kidney injury. Supporting these findings, the overexpression of reduced nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase core subunit V1 (NDUFV1), which encodes a 51-kDa subunit of complex I, can attenuate kidney IR injury by improving mitochondrial function [56].
- Recent studies have revealed that a deficiency in oxidized nicotinamide adenine dinucleotide (NAD+) can contribute to mitochondrial dysfunction, leading to inflammation and kidney disease progression. NAD+ supplementation prevents inflammation during kidney injury. This protective mechanism is attributed to the prevention of mitochondrial RNA leakage into the cytosol and inhibition of cytosolic pattern recognition receptor retinoic acid-inducible gene I (RIG-I), both of which are ameliorated by restoring NAD+ levels [57]. Moreover, it has been shown that NAD+ supplementation can enhance mitochondrial biogenesis in a sirtuin (SIRT)-dependent manner and that its precursor, nicotinamide riboside, can attenuate IR injury [58]. Mitoquinone, a mitochondria-targeted antioxidant, alleviates IR injury by activating the SIRT3-dependent pathway [59]. These findings are supported by a recent study showing that SIRT3 deficiency can dampen early-stage fibrosis after IR injury [60]. The pregnane X receptor (PXR)/aldo-keto reductase family 1, member B7 (AKR1B7) axis was recently introduced as a novel therapeutic target [61]. Another recent study highlighted that peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a master regulator of mitochondrial biogenesis, is downregulated in IR injury [62]. That study further demonstrated that inhibiting forkhead box protein O1 (FOXO1) could restore PGC-1α transcription, implicating the FOXO1 inhibitor AS1842856 as a potential therapeutic agent for IR injury [62].
- Certain drugs have demonstrated promising results. Lasmiditan, a 5-hydroxytryptamine receptor 1F (5-HT1F) agonist, attenuates IR injury by enhancing mitochondrial biogenesis [63]. The importance of 5-HT1F has also been highlighted in a genetic KO model [64]. The antidiabetic drug saxagliptin attenuates IR injury by activating the NRF2/HO-1 pathway [65]. Eplerenone, a mineralocorticoid antagonist available in clinical practice, can reduce kidney IR injury by modulating inflammation and the SIRT1/SIRT3/PGC-1α pathway [66]. Mefunidone, a novel pyridinone drug, can regulate the mitochondria-related Bax/Bcl-2/cleaved-caspase 3 apoptotic pathway, protect mitochondrial electron transport chain complexes III and V levels, and ameliorate kidney function in both in vivo and in vitro disease models [67]. Additionally, acetate, a well-known short-chain fatty acid, reduces cellular ROS production and the cells positive for specific indicator for mitochondrial superoxide (MitoSox, Thermo Fisher Scientific, Waltham, MA, USA) that are indicative of mitochondrial ROS production. Acetate also reduced mitochondrial fission and alleviated IR injury in a murine model [68]. Notably, the ROS-responsive chitosan-SS31 prodrug, which has antioxidant properties, alleviated ROS levels and improved kidney function in a murine model of IR injury [69]. As a member of the vitamin E family, γ-tocotrienol improved mitochondrial function, promoted tubular regeneration, and ameliorated kidney functions in a murine IR model [70]. Treprostinil, a U.S. Food and Drug Administration-approved prostacyclin (PGI2) analog, can improve mitochondrial function and reduce IR injury by a PGC-1α- and SIRT-dependent mechanism [71].
- The aforementioned mPTP opening resulting from mitochondrial dysfunction [10] is also gaining attention as a molecular target for IR injury. A recent study highlighted that mitochondrial ribosomal protein L7/L12 (MRPL12) can bind to adenosine nucleotide translocase 3 (ANT3), thereby stabilizing the mPTP. However, during AKI, MRPL12 expression is reduced in TECs, leading to ANT3 conformational changes and mPTP opening. Overexpression of MRPL12 can protect TECs from apoptosis during hypoxia/reoxygenation challenge [72]. Similarly, Bax inhibitor-1, which conveys anti-apoptotic signals to the mitochondria, is implicated in both IR injury and mitochondrial health [73]. Glutamine administration attenuated kidney damage in vivo during AKI and restored TEC viability in vitro by affecting glutamine gamma glutamyltransferase 2 (Tgm2) and apoptosis signal-regulating kinase (Ask1) [74].
- 2. Targeting mitochondrial dynamics and mitophagy
- Excessive mitochondrial fragmentation has been observed in kidney IR injury [6,9]. There is growing interest in whether strategies to reverse mitochondrial fragmentation can mitigate kidney injury. Recently, it was revealed that the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), known for its functional relationship with cancer and aging, can directly phosphorylate mitochondrial fission 1 protein (Fis1) to induce mitochondrial fission and fragmentation in TECs upon IR. The authors showed that knock-in mice expressing a nonphosphorylatable mutant exhibited improved kidney function, improved histological features, and reduced mitochondrial fragmentation upon AKI induction [75]. Furthermore, SIRT3 promotes mitofusin 2 ubiquitination and degradation, thus suppressing IR-induced AKI [76]. Empagliflozin, an inhibitor of sodium-glucose cotransporter 2, enhanced mitochondrial fusion by promoting the AMPK-OPA1 pathway in an IR model [77]. Emodin (1,3,8-trihydroxy-6-methylanthraquinone), an anthraquinone derivative, demonstrated protective effects in IR-injured mice by suppressing calcium/calmodulin-dependent protein kinase II (CAMKII)/Drp1-mediated mitochondrial fission [78]. Additionally, SIRT3 has been found to alleviate kidney IR injury by enhancing mitochondrial fusion and activating the OPA1 signaling pathway [79].
- Regarding mitophagy, overexpression of BNIP3 is sufficient to overcome kidney IR injury [44,80]. Mesenchymal stem cell-derived extracellular vesicles containing miR-223-3p can activate mitophagy and improve kidney IR injury by inhibiting NLRP3 [81]. Collectively, these findings highlight the potential of mitophagy as a viable target for the treatment of IR injury. Further studies are required to validate the role of small molecules and other compounds that target mitophagy.
- 3. Targeting metabolic reprogramming
- Succinate accumulation during ischemia leads to excessive ROS generation and has been identified as a critical factor in kidney IR injury [82]. Recently, in silico analysis revealed that mitochondrial FAO, peroxisomal lipid metabolism, fatty acid metabolism, and glycolysis are downregulated, whereas the pentose phosphate pathway is upregulated in ischemic kidney tissues [83]. However, the causal relationship between this metabolic reprogramming and its effects requires further investigation. As previously mentioned, PDH activation can diminish succinate accumulation, partly through the restoration of mitochondrial function and attenuation of the malate-aspartate shuttle [9]. Enhanced FAO mediated by carnitine palmitoyltransferase 1A (CPT1A) and delivery of hypoxic mesenchymal stem cell-derived extracellular vesicles can attenuate IR injury, suggesting that metabolic reprogramming is a possible treatment for IR injury [84]. Moreover, AKT serine/threonine kinase 1 (Akt1) and protein kinase B, the main downstream molecules of the insulin-phosphoinositide 3-kinase (PI3K) signaling pathway, are activated during IR injury, dampening tubular apoptosis and kidney injury, which can eventually lead to kidney fibrosis [85,86]. Given that excessive insulin-PI3K-Akt is indicative of insulin resistance, further studies are needed to examine whether improving metabolic conditions, such as insulin sensitization, could ameliorate kidney IR injury.
- 4. Targeting ferroptosis and inflammation
- A recent review provided a comprehensive overview of the role of ferroptosis in kidney IR injury after kidney transplantation [87]. Hence, this section briefly discusses recent therapeutic approaches targeting ferroptosis. Visomitin, a novel mitochondria-targeting antioxidant, can mitigate mitochondrial ROS production, resulting in decreased levels of lipid peroxidation and ferroptosis, thereby protecting against ischemia- or nephrotoxicity-induced AKI [88]. Irisin, an exercise-induced hormone known to improve mitochondrial function and reduce ROS production [89], has been shown to protect against IR injury by upregulating GPX4 in vivo. Importantly, the protective effect of irisin was abrogated by the inhibition of GPX4 [90]. This is supported by an important observation that the ferroptosis suppressor liproxstatin-1 can reduce kidney IR injury and decrease the mortality rate in Gpx4-deficient mice [91].
- Another facet of the mitochondrial contribution to the pathogenesis of IR injury is its relationship with inflammation [92]. Pathogen-associated molecular patterns and damage-associated molecular patterns are important inducers of NLRP3 inflammasome formation in immune cells. Because mitochondrial ROS are among the most important inducers of the NLRP3 inflammasome [92], targeting this process is crucial for addressing IR injury [93]. Remdesivir, a well-known antiviral agent approved for treating coronavirus disease 2019, has been shown to alleviate AKI through NLRP3 inflammasome inhibition [94]. Similarly, the dopamine D1 receptor agonist A68930 can attenuate AKI by inhibiting NLRP3 inflammasome activation [95]. Further research is warranted to understand this novel compound, which targets inflammatory immune cells in addition to macrophages and monocytes, and to develop novel compounds targeting the NLRP3 inflammasome.
Therapeutic strategies of kidney ischemia-reperfusion injury targeting mitochondria
- This review is a comprehensive overview of the interplay between the mitochondria and acute kidney IR injury and provides important insights gleaned from previous studies. Several key points have emerged.
- During acute kidney ischemia, the mitochondria exhibit reduced ATP production and aberrant mitochondrial dynamics, leading to energy depletion, which can adversely affect the survival and function of kidney cells. During the reperfusion period, succinate accumulated during ischemia is rapidly oxidized, releasing electrons toward electron complex I in a process known as reverse electron transport. This abnormal phenomenon results in a ROS burst, triggering the mitochondrial death pathway. Various strategies have been explored to protect the mitochondria, including the use of antioxidants, mitochondria-specific drugs, and interventions to improve mitochondrial metabolism and reduce succinate accumulation.
- Furthermore, mitochondrial damage can trigger inflammatory responses, exacerbating AKI severity. Additionally, mitochondrial dysfunction and the depletion of mitochondrial GPX4 accompany ferroptosis, which occurs during IR injury. Thus, mitochondria also play crucial roles in the regulation of cell death and injury-related inflammation.
- In conclusion, the role of mitochondria in acute kidney IR injury raises several unresolved questions. Therefore, further studies are warranted. The development of novel strategies to effectively safeguard mitochondria and prevent or treat AKI is of paramount importance. Such investigations hold promise for pioneering new directions for the prevention and treatment of acute kidney IR injury.
Conclusion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Funding
This research was supported by Kyungpook National University Development Project Research Fund, 2020.
-
Author contributions
Conceptualization: all authors; Formal analysis, Funding acquisition, Investigation, Supervision: CWH, JHJ; Validation: CWH; Visualization: MJK, CJO; Writing-original draft: MJK, CJO, JHJ; Writing-review & editing: all authors.
Article information



- 1. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 2012;120:c179–84.ArticlePubMedPDF
- 2. Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol 2014;10:193–207.ArticlePubMedPDF
- 3. Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med 2011;17:1391–401.ArticlePubMedPDF
- 4. Sabouny R, Shutt TE. The role of mitochondrial dynamics in mtDNA maintenance. J Cell Sci 2021;134:jcs258944.ArticlePubMedPDF
- 5. Javadov S, Kozlov AV, Camara AK. Mitochondria in health and diseases. Cells 2020;9:1177.ArticlePubMedPMC
- 6. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009;119:1275–85.ArticlePubMedPMC
- 7. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 2017;13:629–46.ArticlePubMedPMCPDF
- 8. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014;515:431–5.ArticlePubMedPMC
- 9. Oh CJ, Kim MJ, Lee JM, Kim DH, Kim IY, Park S, et al. Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia-reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion. Kidney Int 2023;104:724–39.ArticlePubMed
- 10. Nourbakhsh N, Singh P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin Pract 2014;127:149–52.ArticlePubMedPDF
- 11. Yamamoto S, Yamamoto M, Nakamura J, Mii A, Yamamoto S, Takahashi M, et al. Spatiotemporal ATP dynamics during AKI predict renal prognosis. J Am Soc Nephrol 2020;31:2855–69.ArticlePubMedPMC
- 12. Nieuwenhuijs-Moeke GJ, Pischke SE, Berger SP, Sanders JS, Pol RA, Struys MM, et al. Ischemia and reperfusion injury in kidney transplantation: relevant mechanisms in injury and repair. J Clin Med 2020;9:253.ArticlePubMedPMC
- 13. Salvadori M, Rosso G, Bertoni E. Update on ischemia-reperfusion injury in kidney transplantation: pathogenesis and treatment. World J Transplant 2015;5:52–67.ArticlePubMedPMC
- 14. Park JS, Pasupulati R, Feldkamp T, Roeser NF, Weinberg JM. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol 2011;301:F134–50.ArticlePubMedPMC
- 15. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet 2009;43:95–118.ArticlePubMedPMC
- 16. Zhang J, Zhang J, Ni H, Wang Y, Katwal G, Zhao Y, et al. Downregulation of XBP1 protects kidney against ischemia-reperfusion injury via suppressing HRD1-mediated NRF2 ubiquitylation. Cell Death Discov 2021;7:44.ArticlePubMedPMCPDF
- 17. Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, et al. Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov 2023;13:410–31.ArticlePubMedPDF
- 18. Rocca C, Soda T, De Francesco EM, Fiorillo M, Moccia F, Viglietto G, et al. Mitochondrial dysfunction at the crossroad of cardiovascular diseases and cancer. J Transl Med 2023;21:635.ArticlePubMedPMCPDF
- 19. Ma H, Guo X, Cui S, Wu Y, Zhang Y, Shen X, et al. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int 2022;101:315–30.ArticlePubMed
- 20. Yang X, Kang A, Lu Y, Li Y, Guo L, Li R, et al. Exploratory metabolomic analysis based on UHPLC-Q-TOF-MS/MS to study hypoxia-reoxygenation energy metabolic alterations in HK-2 cells. Ren Fail 2023;45:2186715.ArticlePubMedPMC
- 21. Singh P. Reprogramming of energy metabolism in kidney disease. Nephron 2023;147:61–4.ArticlePDF
- 22. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 2016;27:3356–67.ArticlePubMedPMC
- 23. Zhang J, Wang YT, Miller JH, Day MM, Munger JC, Brookes PS. Accumulation of succinate in cardiac ischemia primarily occurs via canonical krebs cycle activity. Cell Rep 2018;23:2617–28.ArticlePubMedPMC
- 24. Spinelli JB, Rosen PC, Sprenger HG, Puszynska AM, Mann JL, Roessler JM, et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science 2021;374:1227–37.ArticlePubMedPMC
- 25. Beach TE, Prag HA, Pala L, Logan A, Huang MM, Gruszczyk AV, et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol 2020;36:101640.ArticlePubMedPMC
- 26. Archer SL. Mitochondrial dynamics: mitochondrial fission and fusion in human diseases. N Engl J Med 2013;369:2236–51.ArticlePubMed
- 27. Xiao X, Hu Y, Quirós PM, Wei Q, López-Otín C, Dong Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am J Physiol Renal Physiol 2014;306:F1318–26.ArticlePubMedPMC
- 28. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 2013;83:568–81.ArticlePubMedPMC
- 29. McBride H, Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr Biol 2010;20:R274–6.ArticlePubMed
- 30. Liu Z, Li H, Su J, Xu S, Zhu F, Ai J, et al. Numb depletion promotes Drp1-mediated mitochondrial fission and exacerbates mitochondrial fragmentation and dysfunction in acute kidney injury. Antioxid Redox Signal 2019;30:1797–816.ArticlePubMed
- 31. Cho SG, Xiao X, Wang S, Gao H, Rafikov R, Black S, et al. Bif-1 Interacts with prohibitin-2 to regulate mitochondrial inner membrane during cell stress and apoptosis. J Am Soc Nephrol 2019;30:1174–91.ArticlePubMedPMC
- 32. Qin N, Cai T, Ke Q, Yuan Q, Luo J, Mao X, et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J Pathol 2019;247:392–405.ArticlePubMedPDF
- 33. Su CT, See DH, Huang YJ, Jao TM, Liu SY, Chou CY, et al. LTBP4 protects against renal fibrosis via mitochondrial and vascular impacts. Circ Res 2023;133:71–85.ArticlePubMed
- 34. Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 2020;15:20.ArticlePubMedPMCPDF
- 35. Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 2015;29:989–99.ArticlePubMedPMC
- 36. Rüb C, Wilkening A, Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res 2017;367:111–23.ArticlePubMedPDF
- 37. Grenier K, McLelland GL, Fon EA. Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front Neurol 2013;4:100.ArticlePubMedPMC
- 38. Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018;14:880–97.ArticlePubMedPMC
- 39. Li N, Wang H, Jiang C, Zhang M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp Cell Res 2018;369:27–33.ArticlePubMed
- 40. Livingston MJ, Wang J, Zhou J, Wu G, Ganley IG, Hill JA, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019;15:2142–62.ArticlePubMedPMCPDF
- 41. Su L, Zhang J, Wang J, Wang X, Cao E, Yang C, et al. Pannexin 1 targets mitophagy to mediate renal ischemia/reperfusion injury. Commun Biol 2023;6:889.ArticlePubMedPMCPDF
- 42. Feng J, Li H, Zhang Y, Wang Q, Zhao S, Meng P, et al. Mammalian STE20-like kinase 1 deletion alleviates renal ischaemia-reperfusion injury via modulating mitophagy and the AMPK-YAP signalling pathway. Cell Physiol Biochem 2018;51:2359–76.ArticlePubMedPDF
- 43. Shi H, Qi H, Xie D, Zhuang J, Qi H, Dai Y, et al. Inhibition of ACSF2 protects against renal ischemia/reperfusion injury via mediating mitophagy in proximal tubular cells. Free Radic Biol Med 2023;198:68–82.ArticlePubMed
- 44. Tang C, Han H, Liu Z, Liu Y, Yin L, Cai J, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis 2019;10:677.ArticlePubMedPMCPDF
- 45. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72.ArticlePubMedPMC
- 46. Hosohata K, Harnsirikarn T, Chokesuwattanaskul S. Ferroptosis: a potential therapeutic target in acute kidney injury. Int J Mol Sci 2022;23:6583.ArticlePubMedPMC
- 47. Eleftheriadis T, Pissas G, Filippidis G, Liakopoulos V, Stefanidis I. Reoxygenation induces reactive oxygen species production and ferroptosis in renal tubular epithelial cells by activating aryl hydrocarbon receptor. Mol Med Rep 2021;23:41.ArticlePubMed
- 48. Eleftheriadis T, Pissas G, Golfinopoulos S, Liakopoulos V, Stefanidis I. Role of indoleamine 2,3-dioxygenase in ischemia-reperfusion injury of renal tubular epithelial cells. Mol Med Rep 2021;23:472.ArticlePubMedPMC
- 49. Sun X, Huang N, Li P, Dong X, Yang J, Zhang X, et al. TRIM21 ubiquitylates GPX4 and promotes ferroptosis to aggravate ischemia/reperfusion-induced acute kidney injury. Life Sci 2023;321:121608.ArticlePubMed
- 50. Liang Y, Liu Z, Qu L, Wang Y, Zhou Y, Liang L, et al. Inhibition of the IRE1/JNK pathway in renal tubular epithelial cells attenuates ferroptosis in acute kidney injury. Front Pharmacol 2022;13:927641.ArticlePubMedPMC
- 51. Tao W, Liu F, Zhang J, Fu S, Zhan H, Qian K. miR-3587 Inhibitor attenuates ferroptosis following renal ischemia-reperfusion through HO-1. Front Mol Biosci 2021;8:789927.ArticlePubMed
- 52. Corridon PR. Enhancing the expression of a key mitochondrial enzyme at the inception of ischemia-reperfusion injury can boost recovery and halt the progression of acute kidney injury. Front Physiol 2023;14:1024238.ArticlePubMedPMC
- 53. Liu Z, Li Y, Li C, Yu L, Chang Y, Qu M. Delivery of coenzyme Q10 with mitochondria-targeted nanocarrier attenuates renal ischemia-reperfusion injury in mice. Mater Sci Eng C Mater Biol Appl 2021;131:112536.ArticlePubMed
- 54. Kubat GB, Kartal Y, Atalay O, Ulger O, Ekinci O, Celik E, et al. Investigation of the effect of isolated mitochondria transplantation on renal ischemia-reperfusion injury in rats. Toxicol Appl Pharmacol 2021;433:115780.ArticlePubMed
- 55. Doulamis IP, Guariento A, Duignan T, Kido T, Orfany A, Saeed MY, et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am J Physiol Renal Physiol 2020;319:F403–13.ArticlePubMedPMC
- 56. Li L, Zhang L, Cao Y, Chen X, Gong H, Ma Y, et al. NDUFV1 attenuates renal ischemia-reperfusion injury by improving mitochondrial homeostasis. J Cell Mol Med 2023;27:1341–52.ArticlePubMedPMC
- 57. Doke T, Mukherjee S, Mukhi D, Dhillon P, Abedini A, Davis JG, et al. NAD+ precursor supplementation prevents mtRNA/RIG-I-dependent inflammation during kidney injury. Nat Metab 2023;5:414–30.ArticlePubMedPMCPDF
- 58. Morevati M, Egstrand S, Nordholm A, Mace ML, Andersen CB, Salmani R, et al. Effect of NAD+ boosting on kidney ischemia-reperfusion injury. PLoS One 2021;16:e0252554.ArticlePubMedPMC
- 59. Mao H, Zhang Y, Xiong Y, Zhu Z, Wang L, Liu X. Mitochondria-Targeted antioxidant mitoquinone maintains mitochondrial homeostasis through the Sirt3-dependent pathway to mitigate oxidative damage caused by renal ischemia/reperfusion. Oxid Med Cell Longev 2022;2022:2213503.ArticlePubMedPMCPDF
- 60. Cheng L, Yang X, Jian Y, Liu J, Ke X, Chen S, et al. SIRT3 deficiency exacerbates early-stage fibrosis after ischaemia-reperfusion-induced AKI. Cell Signal 2022;93:110284.ArticlePubMed
- 61. Yu X, Xu M, Meng X, Li S, Liu Q, Bai M, et al. Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI. Sci Transl Med 2020;12:eaay7591.ArticlePubMed
- 62. Wang D, Wang Y, Zou X, Shi Y, Liu Q, Huyan T, et al. FOXO1 inhibition prevents renal ischemia-reperfusion injury via cAMP-response element binding protein/PPAR-γ coactivator-1α-mediated mitochondrial biogenesis. Br J Pharmacol 2020;177:432–48.ArticlePubMedPDF
- 63. Hurtado KA, Janda J, Schnellmann RG. Lasmiditan promotes recovery from acute kidney injury through induction of mitochondrial biogenesis. Am J Physiol Renal Physiol 2023;324:F56–63.ArticlePubMed
- 64. Gibbs WS, Collier JB, Morris M, Beeson CC, Megyesi J, Schnellmann RG. 5-HT1F receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am J Physiol Renal Physiol 2018;315:F1119–28.ArticlePubMedPMC
- 65. Tang Y, Leng YF, Wang W, Zhang J, Yuan TL, Wang J. Protective effect of Saxagliptin on diabetic rats with renal ischemia reperfusion injury by targeting oxidative stress and mitochondrial apoptosis pathway through activating Nrf-2/HO-1 signaling. Transpl Immunol 2023;76:101762.ArticlePubMed
- 66. Barati A, Rahbar Saadat Y, Meybodi SM, Nouraei S, Moradi K, Kamrani Moghaddam F, et al. Eplerenone reduces renal ischaemia/reperfusion injury by modulating Klotho, NF-κB and SIRT1/SIRT3/PGC-1α signalling pathways. J Pharm Pharmacol 2023;75:819–27.ArticlePubMedPDF
- 67. Li J, Jiang Y, Dai Q, Yu Y, Lv X, Zhang Y, et al. Protective effects of mefunidone on ischemia-reperfusion injury/folic acid-induced acute kidney injury. Front Pharmacol 2022;13:1043945.ArticlePubMedPMC
- 68. Kawabata C, Hirakawa Y, Inagi R, Nangaku M. Acetate attenuates kidney fibrosis in an oxidative stress-dependent manner. Physiol Rep 2023;11:e15774.ArticlePubMedPMC
- 69. Liu D, Shu G, Jin F, Qi J, Xu X, Du Y, et al. ROS-responsive chitosan-SS31 prodrug for AKI therapy via rapid distribution in the kidney and long-term retention in the renal tubule. Sci Adv 2020;6:eabb7422.ArticlePubMedPMC
- 70. Nowak G, Megyesi J. γ-tocotrienol protects against mitochondrial dysfunction, energy deficits, morphological damage, and decreases in renal functions after renal ischemia. Int J Mol Sci 2021;22:12674.ArticlePubMedPMC
- 71. Ding M, Tolbert E, Birkenbach M, Gohh R, Akhlaghi F, Ghonem NS. Treprostinil reduces mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed Pharmacother 2021;141:111912.ArticlePubMedPMC
- 72. Ji X, Chu L, Su D, Sun J, Song P, Sun S, et al. MRPL12-ANT3 interaction involves in acute kidney injury via regulating MPTP of tubular epithelial cells. iScience 2023;26:106656.ArticlePubMedPMC
- 73. Wang J, Zhu P, Li R, Ren J, Zhang Y, Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics 2020;10:384–97.ArticlePubMedPMC
- 74. Thomas K, Zondler L, Ludwig N, Kardell M, Lüneburg C, Henke K, et al. Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells. JCI Insight 2022;7:e163161.ArticlePubMedPMC
- 75. Wang S, Zhu H, Li R, Mui D, Toan S, Chang X, et al. DNA-PKcs interacts with and phosphorylates Fis1 to induce mitochondrial fragmentation in tubular cells during acute kidney injury. Sci Signal 2022;15:eabh1121.ArticlePubMed
- 76. Shen L, Zhang Q, Tu S, Qin W. SIRT3 mediates mitofusin 2 ubiquitination and degradation to suppress ischemia reperfusion-induced acute kidney injury. Exp Cell Res 2021;408:112861.ArticlePubMed
- 77. Yang W, Li X, He L, Zhu S, Lai S, Zhang X, et al. Empagliflozin improves renal ischemia-reperfusion injury by reducing inflammation and enhancing mitochondrial fusion through AMPK-OPA1 pathway promotion. Cell Mol Biol Lett 2023;28:42.ArticlePubMedPMCPDF
- 78. Wang Y, Liu Q, Cai J, Wu P, Wang D, Shi Y, et al. Emodin prevents renal ischemia-reperfusion injury via suppression of CAMKII/DRP1-mediated mitochondrial fission. Eur J Pharmacol 2022;916:174603.ArticlePubMed
- 79. Wang Q, Xu J, Li X, Liu Z, Han Y, Xu X, et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J Cell Physiol 2019;234:23495–506.ArticlePubMedPDF
- 80. Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 2020;36:101671.ArticlePubMedPMC
- 81. Sun Z, Gao Z, Wu J, Zheng X, Jing S, Wang W. MSC-derived extracellular vesicles activate mitophagy to alleviate renal ischemia/reperfusion injury via the miR-223-3p/NLRP3 axis. Stem Cells Int 2022;2022:6852661.ArticlePubMedPMCPDF
- 82. Kamarauskaite J, Baniene R, Trumbeckas D, Strazdauskas A, Trumbeckaite S. Increased succinate accumulation induces ROS generation in in vivo ischemia/reperfusion-affected rat kidney mitochondria. Biomed Res Int 2020;2020:8855585.ArticlePubMedPMCPDF
- 83. Scantlebery AM, Tammaro A, Mills JD, Rampanelli E, Kors L, Teske GJ, et al. The dysregulation of metabolic pathways and induction of the pentose phosphate pathway in renal ischaemia-reperfusion injury. J Pathol 2021;253:404–14.ArticlePubMedPMCPDF
- 84. Gao Z, Zhang C, Peng F, Chen Q, Zhao Y, Chen L, et al. Hypoxic mesenchymal stem cell-derived extracellular vesicles ameliorate renal fibrosis after ischemia-reperfusion injure by restoring CPT1A mediated fatty acid oxidation. Stem Cell Res Ther 2022;13:191.ArticlePubMedPMCPDF
- 85. Kim IY, Song SH, Seong EY, Lee DW, Bae SS, Lee SB. Akt1 is involved in renal fibrosis and tubular apoptosis in a murine model of acute kidney injury-to-chronic kidney disease transition. Exp Cell Res 2023;424:113509.ArticlePubMed
- 86. Lin HY, Chen Y, Chen YH, Ta AP, Lee HC, MacGregor GR, et al. Tubular mitochondrial AKT1 is activated during ischemia reperfusion injury and has a critical role in predisposition to chronic kidney disease. Kidney Int 2021;99:870–84.ArticlePubMed
- 87. Granata S, Votrico V, Spadaccino F, Catalano V, Netti GS, Ranieri E, et al. Oxidative stress and ischemia/reperfusion injury in kidney transplantation: focus on ferroptosis, mitophagy and new antioxidants. Antioxidants (Basel) 2022;11:769.ArticlePubMedPMC
- 88. Song J, Sheng J, Lei J, Gan W, Yang Y. Mitochondrial targeted antioxidant SKQ1 ameliorates acute kidney injury by inhibiting ferroptosis. Oxid Med Cell Longev 2022;2022:2223957.ArticlePubMedPMCPDF
- 89. Batirel S, Bozaykut P, Mutlu Altundag E, Kartal Ozer N, Mantzoros CS. The effect of Irisin on antioxidant system in liver. Free Radic Biol Med 2014;75(Suppl 1):S16.Article
- 90. Zhang J, Bi J, Ren Y, Du Z, Li T, Wang T, et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. J Cell Physiol 2021;236:931–45.ArticlePubMedPDF
- 91. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014;16:1180–91.ArticlePubMedPMCPDF
- 92. Tschopp J. Mitochondria: sovereign of inflammation? Eur J Immunol 2011;41:1196–202.ArticlePubMed
- 93. Su X, Liu B, Wang S, Wang Y, Zhang Z, Zhou H, et al. NLRP3 inflammasome: a potential therapeutic target to minimize renal ischemia/reperfusion injury during transplantation. Transpl Immunol 2022;75:101718.ArticlePubMed
- 94. Yin L, Zhao H, Zhang H, Li Y, Dong Y, Ju H, et al. Remdesivir alleviates acute kidney injury by inhibiting the activation of NLRP3 inflammasome. Front Immunol 2021;12:652446.ArticlePubMedPMC
- 95. Cao JY, Zhou LT, Li ZL, Yang Y, Liu BC, Liu H. Dopamine D1 receptor agonist A68930 attenuates acute kidney injury by inhibiting NLRP3 inflammasome activation. J Pharmacol Sci 2020;143:226–33.ArticlePubMed
 PubReader
PubReader ePub Link
ePub Link Cite
Cite