E-Submission
E-SubmissionPubMed Central, CAS, DOAJ, KCI

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 40(2); 2023 > Article
-
Review article
Hepatic encephalopathy on magnetic resonance imaging and its uncertain differential diagnoses: a narrative review -
Chun Geun Lim*
 , Myong Hun Hahm*, Hui Joong Lee
, Myong Hun Hahm*, Hui Joong Lee -
Journal of Yeungnam Medical Science 2023;40(2):136-145.
DOI: https://doi.org/10.12701/jyms.2022.00689
Published online: January 10, 2023
Department of Radiology, Kyungpook National University Hospital, School of Medicine, Kyungpook National University, Daegu, Korea
- Corresponding author: Hui Joong Lee, MD, PhD Department of Radiology, Kyungpook National University Hospital, School of Medicine, Kyungpook National University, 130 Dongduk-ro, Jung-gu, Daegu 41944, Korea Tel.: +82-53-420-5397 • Fax: +82-53-422-2677 • E-mail: leehuijoong@knu.ac.kr
- *These authors contributed equally to this work.
Copyright © 2023 Yeungnam University College of Medicine, Yeungnam University Institute of Medical Science
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Hepatic encephalopathy (HE) is a severe neuropsychiatric abnormality in patients with either acute or chronic liver failure. Typical brain magnetic resonance imaging findings of HE are bilateral basal ganglia high signal intensities due to manganese deposition in chronic liver disease and hyperintensity in T2, fluid-attenuated inversion recovery, or diffusion-weighted imaging (DWI) with hemispheric white matter changes including the corticospinal tract. Low values on apparent diffusion coefficient mapping of the affected area on DWI, indicating cytotoxic edema, can be observed in acute HE. However, neuropsychological impairment in HE ranges from mild deficits in psychomotor abilities affecting quality of life to stupor or coma with higher grades of hepatic dysfunction. In particular, the long-lasting compensatory mechanisms for the altered metabolism in chronic liver disease make HE imaging results variable. Therefore, the clinical relevance of imaging findings is uncertain and differentiating HE from other metabolic diseases can be difficult. The recent introduction of concepts such as “acute-on-chronic liver failure (ACLF),” a new clinical entity, has led to a change in the clinical view of HE. Accordingly, there is a need to establish a corresponding concept in the field of neuroimaging diagnosis. Herein, we review HE from a historical and etiological perspective to increase understanding of brain imaging and help establish an imaging approach for advanced new concepts such as ACLF. The purpose of this manuscript is to provide an understanding of HE by reviewing neuroimaging findings based on pathological and clinical concepts of HE, thereby assisting in neuroimaging interpretation.
- Hepatic encephalopathy (HE) is a severe neuropsychiatric abnormality in patients with either acute or chronic liver failure [1,2]. Typical magnetic resonance imaging (MRI) findings of chronic liver failure are high signal intensity in bilateral basal ganglia high signal intensities (BGH) due to manganese deposition, and high signal intensity in fluid-attenuated inversion recovery (FLAIR) and diffusion-weighted imaging (DWI) images with hemispherical white matter changes including the corticospinal tract. In acute HE, low apparent diffusion coefficient (ADC) values in the affected area reflect cytotoxic edema. However, neuropsychological impairment in HE manifests as a spectrum ranging from mild deficits in psychomotor abilities affecting quality of life to stupor or coma with higher grades of hepatic dysfunction [2]. In particular, the long-lasting compensatory mechanism for the altered metabolism in chronic liver disease (CLD) makes HE imaging findings very variable. Therefore, the clinical relevance of imaging findings is questionable, and it is difficult to differentiate HE from other metabolic diseases. The recent acceptance of new clinical concepts such as “ acute-on-chronic liver failure (ACLF)” has led to a change in the clinical view of HE. Accordingly, there is a need to reevaluate existing paradigms in the field of neuroimaging diagnosis. The purpose of this manuscript is to provide a comprehensive review of HE through neuroimaging based on the pathological and clinical understanding of HE, thereby assisting in the interpretation of neuroimaging findings.
Introduction
- Ethical statements: Written informed consent was obtained from the patient for publication of this review article and any accompanying images.
- MRI is a very useful tool to better understand the pathophysiology of HE and provide direct evidence for the pathogenesis of HE due to severe liver dysfunction. In particular, magnetic resonance spectroscopy (MRS) provides information on the pathophysiological changes in HE at the molecular level.
- 1. Basal ganglia high signal intensity
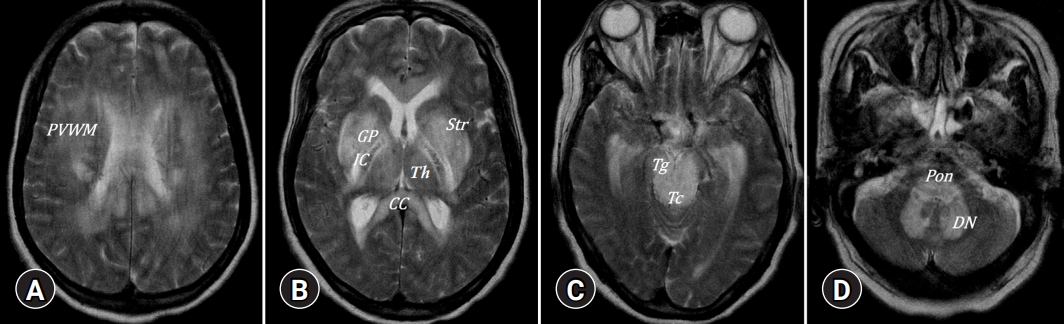
- The most frequent MRI finding in the brains of patients with CLD is BGH on T1-weighted imaging without mass effect, which results from paramagnetic manganese deposition [3-5]. Manganese crosses the blood-brain barrier and accumulates in the globus pallidus in chronic liver failure, which may be the cause of extrapyramidal symptoms [6]. BGH is thought to be caused by the severity of liver dysfunction and is reversible when liver function returns to normal levels after liver transplantation [7]. Manganese plays an essential role in the normal function of several enzymes, including glutamine synthetase and mitochondrial superoxide dismutase. Manganese has a neurotoxic effect, sparing the nigrostriatal system and causing selective neuronal death in the basal ganglia, structures, and reactive gliosis. This cell death is more apparent in the globus pallidus and substantia nigra reticulata, and less apparent in the striatum and bilateral internal capsules (Fig. 1). Clinically, early gait and balance dysfunction, the relative absence of resting tremors, the presence of mild cognitive impairment at the time of presentation, and little or no response to levodopa distinguish Parkinsonian manifestations in HE from idiopathic Parkinson disease. The pallidal index, used as a semiquantitative parameter to evaluate manganese accumulation in the brain, is a relative value calculated as the ratio of the signal intensity in the globus pallidus to the subcortical frontal white matter in axial T1-weighted images. The pallidal index correlates with clinical indices such as whole blood manganese, Child-Pugh score, and total bilirubin level, which reveals the severity of the portal-systemic shunt [8,9].
- 2. White matter involvement
- Due to minor brain edema that is beyond the threshold for detection on conventional MRI, hyperintensity along the white matter in the cerebral hemisphere or near the corticospinal tract on FLAIR or T2-weighted imaging has been described, mimicking the MRI features of amyotrophic lateral sclerosis [10] (Fig. 2). Transcallosal white matter involvement, so-called Marchiafava-Bignami disease (MBD), is a typical form of white matter involvement in patients with HE [11]. It was believed that MBD was specific to those who resided in central Italy and drank a lot of inexpensive red wine from the Chianti region. However, it is now known worldwide that the cause of MBD is closely related to alcohol consumption. The mechanism of white matter involvement in acute HE is not completely understood. The lack of integrity of the transcallosal connections between motor cortices is the consequence of axonal degeneration of transcallosal fibers. According to another hypothesis, arylsulfatase A (ASA) activity is reduced in patients with alcoholic cirrhosis similar to metachromatic leukodystrophy, which alters sphingomyelin metabolism [12].
- Focal white matter T2-weighted lesions (WMLs) may also be observed in liver cirrhosis with or without overt HE. These lesions look like different types of small-vessel disease and white matter hyperintensity in healthy people who are elderly. However, focal WMLs in HE can be partially reversible with HE recovery or after liver transplantation, unlike other WMLs [13].
- 3. Cortical changes
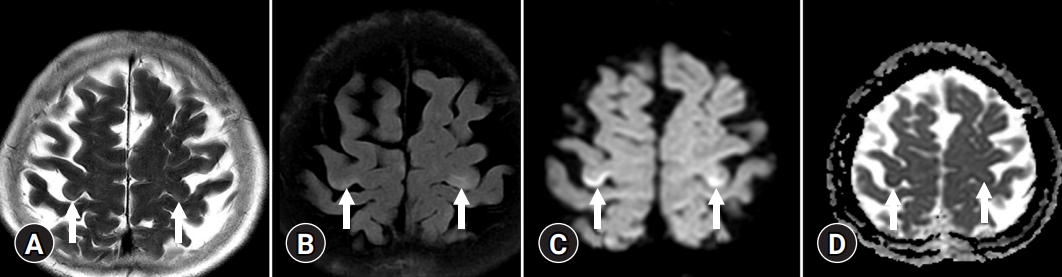
- On T2-weighted and FLAIR images, acute HE is characterized by diffuse cortical, overt brain edema, while cortical-restricted diffusion has a high signal intensity on DWI and low signal intensity on ADC maps, indicating cytotoxic edema (Fig. 3) [14]. The cingulate gyrus and insular cortex were symmetrically involved in the brain MRIs of all patients, with additional cortical involvement being more variable and asymmetrical. However, involvement of the parietal, frontal, temporal, or occipital cortex is unusual [15].
- 4. Magnetic resonance spectroscopy
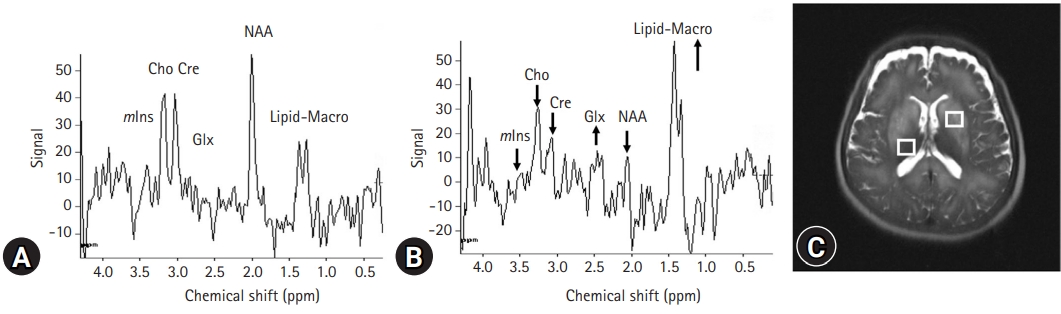
- Currently, there is agreement on the characteristic triad of proton magnetic resonance spectroscopy (1H-MRS) results in HE, including depletion of intracellular choline (Cho) and myoinositol (mIns) as well as accumulation of glutamine (Gln), all of which are associated with neuropsychiatric dysfunction (Fig. 4) [16]. Signals of glutamate and Gln are increased to compensate for glial Gln accumulation to maintain osmotic homeostasis [17]. However, the decompensation of these volume-regulatory mechanisms may result in neuroglial disturbance and astrocyte swelling. Among these 1H-MRS variables, mIns appears to be a more sensitive biomarker in the early detection of HE [18]. Diminished mIns, which contributes to ion flux, may act as an organic osmolyte and is critical for controlling astrocyte volume. Cho is a component of phosphocholine, which is related to cell membrane activation. Cho is a necessary component in the synthesis of acetylcholine, which is a neurotransmitter associated with memory, awareness, and feelings. A reduced ratio of Cho to creatinine may underlie cognitive impairment in HE. In patients with cirrhosis, the majority of MRS studies found no significant changes in N-acetylaspartate, which may indicate no discernible neuronal impairment as HE progressed [19]. In the past, it was believed that neuronal changes in HE were either absent or unimportant in explaining the neuropsychiatric abnormality. However, a neuropathological study subsequently showed significant neuronal cell loss in the brains of patients with HE. Dopaminergic and serotoninergic neuronal systems, as well as Purkinje cells, have also been shown to decline in HE [20].
Typical neuroimaging of hepatic encephalopathy
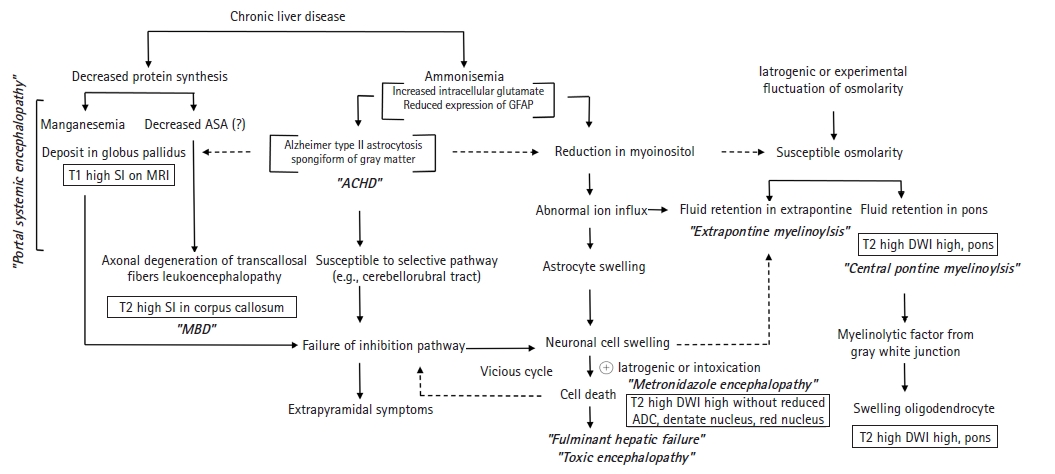
- We have summarized the pathophysiology of HE associated with neuroimaging in Fig. 5. It is essential for radiologists to understand the biological mechanisms to understand this figure. First, it is necessary to understand why patients with HE are vulnerable to osmotic damage and how metabolic changes affect magnetic resonance signals.
- 1. Metabolites
- Ammonia is believed to play an important role in the cause of HE. The liver detoxifies and converts the majority of ammonia absorbed from the small intestine through the portal vein into urea. Thus, HE may result from hepatic dysfunction, defects in urea circulation, or portocaval shunts that increase blood ammonia levels. Skeletal muscle and brain astrocytes contribute to the detoxification of ammonia by converting it to Gln in cases of hepatic dysfunction. Under the catalysis of glutamine synthase, elevated ammonia and Glu are converted to abundant levels of Gln, which contribute to elevated osmotic pressure. Although the mechanism of brain edema in acute hepatic failure is not fully understood, ammonia is generally assumed to play a critical role. In addition, reduced urea cycle activity in chronic liver dysfunction leads to an increase in cerebral Gln synthesis by brain astrocytes [21].
- Hyponatremia causes not only rapid loss of intracellular electrolytes such as potassium but also low-molecular-weight organic osmolytes including mIns and Cho [22]. The decreased mIns contributing to ion flux may serve as an organic osmolyte and plays a more important role in the volume regulation of astrocytes than intracellular electrolytes in such situations [23]. During hyperglycemia in unmanaged diabetes and ethanol intoxication, extracellular osmolality may increase despite normal or decreased extracellular Na+ concentrations. Astrocyte swelling may not be visible until compensatory osmoregulatory mechanisms, such as loss of intracellular osmolytes, disappear. For this reason, there were cases in which Na+ concentration changes related to central pontine myelinolysis (CPM) could not be found in the presence of metabolic diseases. Since the pons and lateral geniculate body appear to be the exclusive anatomical locations associated with osmotic demyelination syndrome, deep gray matter involvement is highly likely to result in the osmotic injury related to acquired chronic hepatocerebral degeneration (ACHD). This is expected to be revealed in a detailed study on whether extrapontine myelinolysis (EPM)/CPM-like lesions occur in patients with ACHD and those with HE.
- 2. Pathology
- Pathologically, HE may be classified into portal-systemic encephalopathy (PSE) and HE in fulminant hepatic failure (FHF) [24]. PSE is a complication of portal-systemic shunting of venous blood, which can develop either spontaneously as a result of portal hypertension or after surgical intervention. Astrocytes, a type of neural cell, are most susceptible to the effects of liver failure [25]. Critical astrocytic proteins, such as the structural glial fibrillary acidic protein (GFAP), a cytoplasmic filamentous protein that makes up a significant portion of the cellular component in mature astrocytes, are downregulated when the brain is exposed to ammonia [26]. Diminished GFAP expression causes morphological changes in astrocytes that favor extracellular space diffusivity. The presence of the astrocytic pathology known as Alzheimer type II astrocytosis, in which astrocytes acquire a characteristic swollen shape with a large pale nucleus, a prominent nucleolus, and margination of the chromatin, was shown in histopathologic studies of brain sections of patients with cirrhosis [27]. At autopsy, a subset of patients with hepatocerebral degeneration may have band-like or patchy central nervous system (CNS) myelin vacuolization that is not related to myelin breakdown or macrophage influx. In nearby astrocytes, typical changes indicative of chronic liver failure were observed [25]. Neurologically, PSE develops slowly; the onset is often insidious starting with personality changes. ACHD occurred in patients with portocaval shunts or liver cirrhosis resulting from a number of causes, but not Wilson disease [25].
- Unlike PSE, cytotoxic edema rather than vasogenic edema is visible in the brain tissue of FHF, according to electron microscopic studies [28]. Characteristically cytotoxic brain edema and especially swelling of astrocytes and astrocytic endfeet, among several brain cell types, have been most frequently observed in acute liver failure [28]. Mortality rates are higher in FHF than in PSE; brainstem herniation caused by increased intracranial pressure as a consequence of brain edema is the most common cause of death. In contrast to PSE, patients with FHF progress through states of altered mental capacity and confusion to stupor and coma within a few hours or days.
- 3. Neurotransmission
- Impairment of neuronal communication in HE can result from changes in numerous neurotransmitter systems. High concentrations of ammonium ions have been shown to obstruct glutamatergic excitatory transmission. Increased serotonin turnover caused by chronic hyperammonemia may be the cause of the altered sleep patterns in HE [29]. Ammonia is also known to damage brain energy metabolism and blood flow autoregulation. Recent studies have shown that ammonia exposure causes cultured astrocytes to undergo a process known as mitochondrial permeability transition, which is linked to mitochondrial failure and subsequent cellular dysfunction and has similar mechanisms to those of Wernicke encephalopathy [30].
- Acute liver failure results in altered expression of several genes in brain, some of which code for proteins such as the glucose (glucose transporter 1) and glutamate (glutamate transporter-1, GLT-1) transporters, the astrocytic structural protein GFAP the “peripheral-type” benzodiazepine receptor and the water channel protein, aquaporin IV. Loss of expression of GLT-1 results in increased extracellular brain glutamate [29]. In chronic liver dysfunction, reduced urea cycle activity causes astrocytes to increase synthesis of cerebral Gln.
Pathophysiology of hepatic encephalopathy
- 1. Classic staging
- There are several clinical classifications and a grading system for HE according to cause and severity. According to the underlying disease, HE is subclassified into type A (acute liver failure), type B (bypass or shunt), and type C (cirrhosis). Type C HE is subcategorized into episodic (acute), recurrent, and persistent HE, depending on the time course. According to the existence of precipitating factors, episodic HE is subdivided into spontaneous (nonprecipitated), precipitated, or recurrent HE (when two episodes of episodic HE occur in 1 year). Precipitating factors can be identified in almost all cases of episodic hepatic type C [31]. Based on the clinical severity from subclinical alterations to severe coma or death, HE may be subclassified by West Haven Criteria (WHC) into grades 0 to IV [32]. To overcome the limited interobserver reliability of the criteria, the International Society for Hepatic Encephalopathy and Nitrogen Metabolism classification was proposed. Patients with minimal HE and WHC grade I would be classified as having covert HE. Other patients with WHC grades II to IV would be classified as having overt HE, a counterpart of covert HE [33].
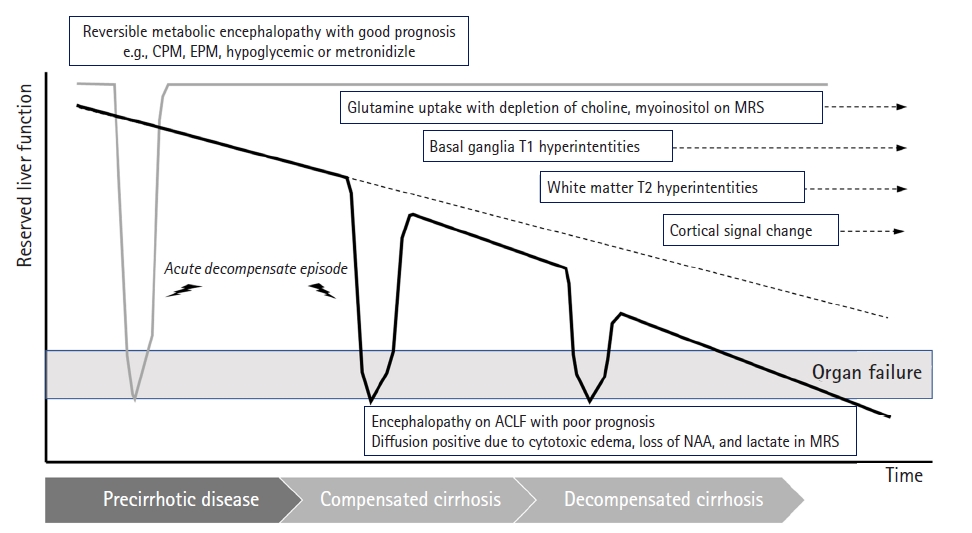
- 2. Acute-on-chronic liver failure
- Acutely decompensated cirrhosis and ACLF share two important clinical features: known CLD with acute decompensation. ACLF, a term proposed by Jalan and Williams [34], originated from studies that revealed a syndrome related to a high risk of short-term death (death <4 weeks after admission) in patients with acutely decompensated cirrhosis. Three major clinical features of ACLF are intense systemic inflammation, frequent association with proinflammatory precipitating events (such as infections or alcoholic hepatitis), and single- or multi-extrahepatic organ failure (e.g., function of kidney, brain, blood coagulation, circulation, and respiration) (Fig. 6) [35,36].
Evolution of clinical staging
- 1. Osmotic demyelination
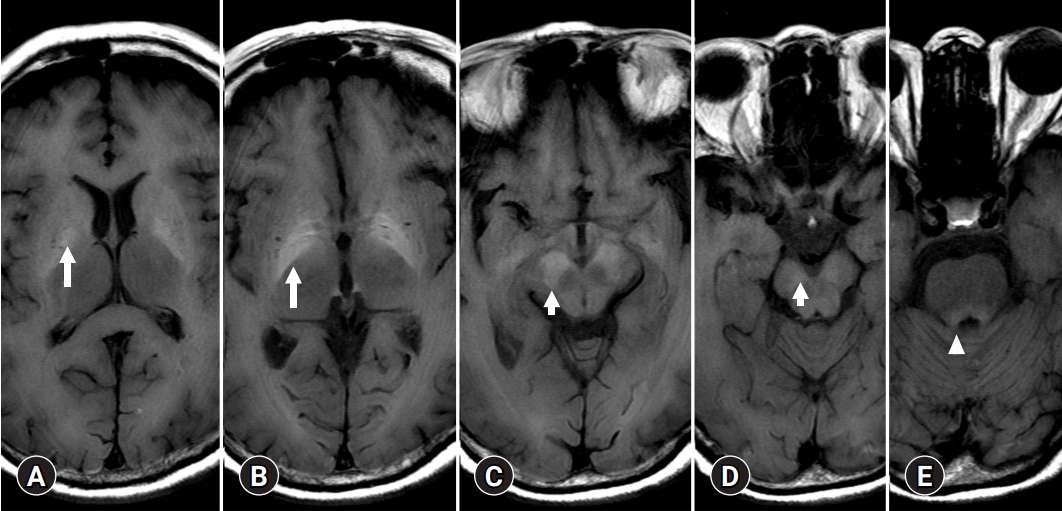
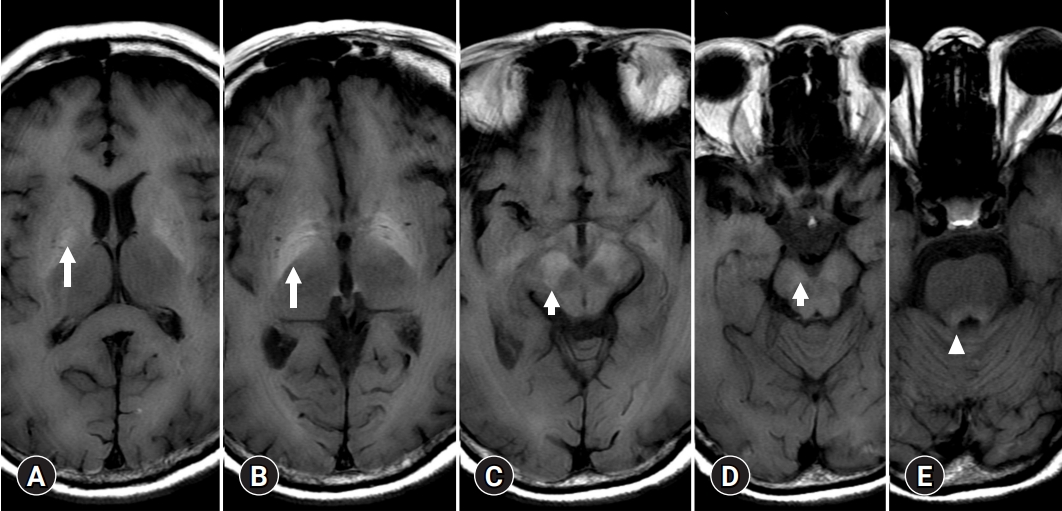
- A few cases of HE show T2 or diffusion high signal intensities of basal ganglia mimicking EPM, or uremic encephalopathy (Fig. 7). HE shares common clinical features with other metabolic diseases such as osmotic demyelination or uremic encephalopathy. There are two possible explanations for the symmetric and synchronous involvement of deep gray matter. One is the concept of ACHD and the other is EPM [37].
- As a CPM, a symmetrical solitary, midline lesion in the basis pontis with myelin breakdown, macrophage influx, relative preservation of axis cylinders, and minimal inflammatory response was first described in 1959 as a “hitherto undescribed disease” [38]. CPM is regarded as a disease occurring in alcoholic and malnourished patients with chronic renal failure or hepatocellular dysfunction [39]. In 58 autopsy cases of CPM, EPM was observed more frequently than a solitary pontine lesion [40]. Through clinical and experimental studies, the onset of CPM and EPM was demonstrated to occur concomitantly with a rapid correction in hyponatremia and osmotic shift. Especially with chronic liver dysfunction, the decreased urea cycle activity causes astrocytes to increase synthesis of cerebral Gln and decrease mIns, which plays an important role in the regulation of astrocytic volume [37]. Therefore, the MRI signal intensity of deep gray matter in patients with HE can be viewed as sensitive to osmotic disease based on organic changes rather than simple osmotic demyelinating disease.
- 2. Deep gray matter and connectivity
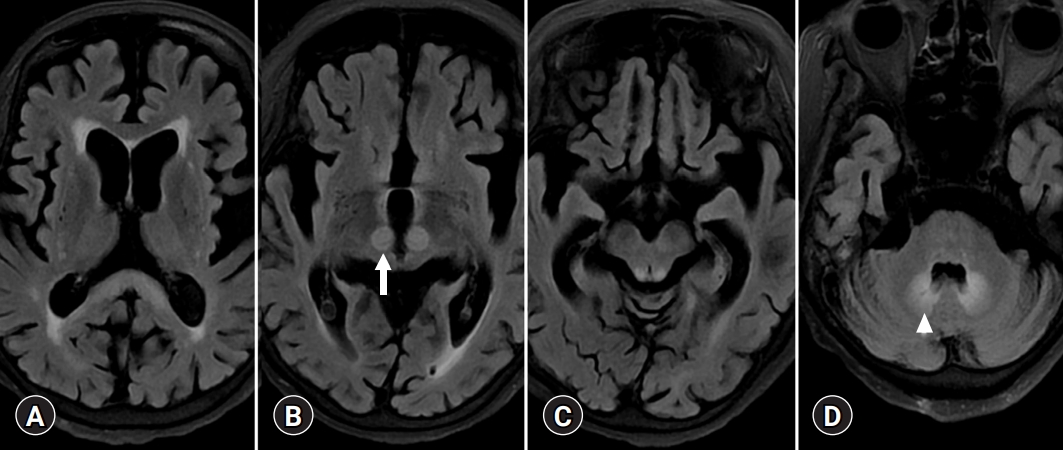
- Bilateral involvement of the dentate nucleus was initially thought to be specific to metronidazole encephalopathy (Fig. 8) [41]. However, many case reports of metronidazole encephalopathy reported synchronous involvement of the red nucleus and dentate nucleus [41], which are components of the Guillain-Mollaret triangle [42]. In addition, there may be bilateral T2 intensities in the dentate nucleus in HE related to methyl bromide poisoning, enteroviral encephalomyelitis, and maple syrup urine disease [43-45]. Therefore, there is a limit to associating the cause of metabolic brain disease with the onset of specific anatomical lesions, and understanding the functional connection between various brain responses and toxic substances is necessary.
Uncertain differential diagnoses
- We used this review to clarify the similarities and differences between HE and lesions belonging to other similar disease entities and to address the uncertainties surrounding their potentially overlapping features by scrutinizing the literature, especially the original descriptions of these entities. We hypothesized that disease progression with varying degrees of corpus callosum and deep cerebral gray matter involvement and the worsening of HE conceptually overlapped. However, EPM and ACHD are pathologically different. EPM may show severe demyelination and macrophage-mediated destruction, while ACHD shows band-shaped or speckled vacuolization of CNS myelin without demyelination or macrophage influx. Therefore, the neuropsychiatric abnormality of HE originates from a combination of several synergistic facilitating factors, and CLD induces a state sensitive to injury from osmotic imbalances or external toxic substances through changes in ACHD, resulting in brain lesions similar to those diseases, such as osmotic demyelination syndrome or toxic encephalopathy. To understand the pathophysiological mechanism of HE, multimodality MRI is a useful and practical research tool. It will also become increasingly vital in the early diagnosis, prognosis, and monitoring of HE.
Conclusion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Funding
None.
-
Author contributions
Conceptualization, Resources: all authors; Data curation: CGL, MHH; Formal analysis: CGL, HJL; Investigation, Software, Validation: MHH; Supervision: HJL; Writing-original draft: CGL, MHH; Writing-review & editing: HJL.
Notes


- 1. Hazell AS, Butterworth RF. Hepatic encephalopathy: an update of pathophysiologic mechanisms. Proc Soc Exp Biol Med 1999;222:99–112.ArticlePubMed
- 2. Sherlock S. Hepatic encephalopathy. Br J Hosp Med 1977;17:144-6, 151-4, 159.ArticlePubMed
- 3. Inoue E, Hori S, Narumi Y, Fujita M, Kuriyama K, Kadota T, et al. Portal-systemic encephalopathy: presence of basal ganglia lesions with high signal intensity on MR images. Radiology 1991;179:551–5.ArticlePubMed
- 4. Syh HW, Chu WK, Mar N, McConnell JR. An image analysis on MR imaging of the brain for hepatic encephalopathy. Biomed Sci Instrum 1991;27:29–33.PubMed
- 5. Zeneroli ML, Cioni G, Crisi G, Vezzelli C, Ventura E. Globus pallidus alterations and brain atrophy in liver cirrhosis patients with encephalopathy: an MR imaging study. Magn Reson Imaging 1991;9:295–302.ArticlePubMed
- 6. Zerón HM, Rodríguez MR, Montes S, Castañeda CR. Blood manganese levels in patients with hepatic encephalopathy. J Trace Elem Med Biol 2011;25:225–9.ArticlePubMed
- 7. Pujol A, Pujol J, Graus F, Rimola A, Peri J, Mercader JM, et al. Hyperintense globus pallidus on T1-weighted MRI in cirrhotic patients is associated with severity of liver failure. Neurology 1993;43:65–9.ArticlePubMed
- 8. Krieger D, Krieger S, Jansen O, Gass P, Theilmann L, Lichtnecker H. Manganese and chronic hepatic encephalopathy. Lancet 1995;346:270–4.ArticlePubMed
- 9. Lee DH, Lee HJ, Hahm MH. The pallidal index in patients with acute-on-chronic liver disease: is it a predictor of severe hepatic encephalopathy? Investig Magn Reson Imaging 2017;21:125–30.ArticlePDF
- 10. Rovira A, Córdoba J, Sanpedro F, Grivé E, Rovira-Gols A, Alonso J. Normalization of T2 signal abnormalities in hemispheric white matter with liver transplant. Neurology 2002;59:335–41.ArticlePubMed
- 11. Gabriel S, Grossmann A, Höppner J, Benecke R, Rolfs A. Marchiafava-Bignami syndrome: extrapontine myelinolysis in chronic alcoholism. Nervenarzt 1999;70:349–56.ArticlePubMed
- 12. Alvarez-Leal M, Contreras-Hernández D, Chávez A, Diaz-Contreras JA, Careaga-Olivares J, Zúñiga-Charles MA, et al. Leukocyte arylsulfatase A activity in patients with alcohol-related cirrhosis. Am J Hum Biol 2001;13:297–300.ArticlePubMed
- 13. Rovira A, Alonso J, Córdoba J. MR imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol 2008;29:1612–21.ArticlePubMedPMC
- 14. Arnold SM, Els T, Spreer J, Schumacher M. Acute hepatic encephalopathy with diffuse cortical lesions. Neuroradiology 2001;43:551–4.ArticlePubMedPDF
- 15. Matsusue E, Kinoshita T, Ohama E, Ogawa T. Cerebral cortical and white matter lesions in chronic hepatic encephalopathy: MR-pathologic correlations. AJNR Am J Neuroradiol 2005;26:347–51.PubMedPMC
- 16. Kreis R, Ross BD, Farrow NA, Ackerman Z. Metabolic disorders of the brain in chronic hepatic encephalopathy detected with H-1 MR spectroscopy. Radiology 1992;182:19–27.ArticlePubMed
- 17. Binesh N, Huda A, Thomas MA, Wyckoff N, Bugbee M, Han S, et al. Hepatic encephalopathy: a neurochemical, neuroanatomical, and neuropsychological study. J Appl Clin Med Phys 2006;7:86–96.Article
- 18. Binesh N, Huda A, Bugbee M, Gupta R, Rasgon N, Kumar A, et al. Adding another spectral dimension to 1H magnetic resonance spectroscopy of hepatic encephalopathy. J Magn Reson Imaging 2005;21:398–405.ArticlePubMed
- 19. Taylor-Robinson SD, Sargentoni J, Oatridge A, Bryant DJ, Hajnal JV, Marcus CD, et al. MR imaging and spectroscopy of the basal ganglia in chronic liver disease: correlation of T1-weighted contrast measurements with abnormalities in proton and phosphorus-31 MR spectra. Metab Brain Dis 1996;11:249–68.ArticlePubMedPDF
- 20. Butterworth R. Neuronal cell death in hepatic encephalopathy. Metab Brain Dis 2007;22:309–20.ArticlePubMedPDF
- 21. Norenberg MD, Bender AS. Astrocyte swelling in liver failure: role of glutamine and benzodiazepines. Acta Neurochir Suppl (Wien) 1994;60:24–7.ArticlePubMed
- 22. Kril JJ, Butterworth RF. Diencephalic and cerebellar pathology in alcoholic and nonalcoholic patients with end-stage liver disease. Hepatology 1997;26:837–41.ArticlePubMed
- 23. Kreis R, Farrow N, Ross BD. Localized 1H NMR spectroscopy in patients with chronic hepatic encephalopathy: analysis of changes in cerebral glutamine, choline and inositols. NMR Biomed 1991;4:109–16.ArticlePubMed
- 24. Romero-Gómez M, Montagnese S, Jalan R. Hepatic encephalopathy in patients with acute decompensation of cirrhosis and acute-on-chronic liver failure. J Hepatol 2015;62:437–47.ArticlePubMed
- 25. Victor M, Adams RD, Cole M. The acquired (non-Wilsonian) type of chronic hepatocerebral degeneration. Medicine (Baltimore) 1965;44:345–96.ArticlePubMed
- 26. Sobel RA, DeArmond SJ, Forno LS, Eng LF. Glial fibrillary acidic protein in hepatic encephalopathy: an immunohistochemical study. J Neuropathol Exp Neurol 1981;40:625–32.ArticlePubMed
- 27. Butterworth RF, Giguère JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol 1987;6:1–12.ArticlePDF
- 28. Kato M, Hughes RD, Keays RT, Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology 1992;15:1060–6.ArticlePubMed
- 29. Desjardins P, Bélanger M, Butterworth RF. Alterations in expression of genes coding for key astrocytic proteins in acute liver failure. J Neurosci Res 2001;66:967–71.ArticlePubMed
- 30. Rama Rao KV, Chen M, Simard JM, Norenberg MD. Suppression of ammonia-induced astrocyte swelling by cyclosporin A. J Neurosci Res 2003;74:891–7.ArticlePubMed
- 31. Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy--definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002;35:716–21.ArticlePubMedPDF
- 32. Ridola L, Faccioli J, Nardelli S, Gioia S, Riggio O. Hepatic encephalopathy: diagnosis and management. J Transl Int Med 2020;8:210–9.ArticlePubMedPMC
- 33. Häussinger D, Schliess F, Kircheis G. Pathogenesis of hepatic encephalopathy. J Gastroenterol Hepatol 2002;17(Suppl 3):S256–9.ArticlePubMedPDF
- 34. Jalan R, Williams R. Acute-on-chronic liver failure: pathophysiological basis of therapeutic options. Blood Purif 2002;20:252–61.ArticlePubMedPDF
- 35. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013;144:1426–37.ArticlePubMed
- 36. Arroyo V, Moreau R, Jalan R. Acute-on-chronic liver failure. N Engl J Med 2020;382:2137–45.ArticlePubMed
- 37. Kleinschmidt-DeMasters BK, Filley CM, Rojiani AM. Overlapping features of extrapontine myelinolysis and acquired chronic (non-Wilsonian) hepatocerebral degeneration. Acta Neuropathol 2006;112:605–16.ArticlePubMedPDF
- 38. Adams RD, Victor M, Mancall EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry 1959;81:154–72.ArticlePubMed
- 39. Ho VB, Fitz CR, Yoder CC, Geyer CA. Resolving MR features in osmotic myelinolysis (central pontine and extrapontine myelinolysis). AJNR Am J Neuroradiol 1993;14:163–7.PubMedPMC
- 40. Gocht A, Colmant HJ. Central pontine and extrapontine myelinolysis: a report of 58 cases. Clin Neuropathol 1987;6:262–70.PubMed
- 41. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol 2007;28:1652–8.ArticlePubMedPMC
- 42. Behzadi F, Fiester PJ, Rao D. Bilateral hypertrophic olivary degeneration following brainstem insult: a retrospective review and examination of causative pathology. Neurosci Insights 2021;16:26331055211007445.ArticlePubMedPMCPDF
- 43. Jang S, Suh SI, Ha SM, Byeon JH, Eun BL, Lee YH, et al. Enterovirus 71-related encephalomyelitis: usual and unusual magnetic resonance imaging findings. Neuroradiology 2012;54:239–45.ArticlePubMedPDF
- 44. Di Rocco M, Biancheri R, Rossi A, Allegri AE, Vecchi V, Tortori-Donati P. MRI in acute intermittent maple syrup urine disease. Neurology 2004;63:1078.ArticlePubMed
- 45. Geyer HL, Schaumburg HH, Herskovitz S. Methyl bromide intoxication causes reversible symmetric brainstem and cerebellar MRI lesions. Neurology 2005;64:1279–81.ArticlePubMed
References
Figure & Data
References
Citations
- Pattern Clustering of Symmetric Regional Cerebral Edema on Brain MRI in Patients with Hepatic Encephalopathy
Chun Geun Lim, Hui Joong Lee
Journal of the Korean Society of Radiology.2024; 85(2): 381. CrossRef - Rule out all differential causes before attributing cerebral bleeding to 5-aminolevulinic acid
Josef Finsterer, Sounira Mehri
Child's Nervous System.2023; 39(4): 847. CrossRef - Minimal hepatic encephalopathy: clinical, neurophysiological, neuroimaging markers
P. I. Kuznetsova, A. A. Raskurazhev, S. N. Morozova, I. M. Lovchev, M. S. Novruzbekov, M. M. Tanashyan
Russian neurological journal.2023; 28(5): 21. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite