Anti-nuclear antibody-negative immunoglobulin G4-associated autoimmune hepatitis mimicking lymphoproliferative disorders
Article information

Abstract
Immunoglobulin G4 (IgG4)-associated autoimmune hepatitis (AIH) is a very rare subtype of autoimmune hepatitis and characterized by marked elevated serum IgG and hepatic infiltration of IgG4-expressing plasma cells. Pathologic confirmation of hepatic IgG4-expressing plasma cells is usually required for the final diagnosis of IgG4-associated AIH. Herein, we report the case of a 47-year-old female diagnosed with autoantibody-negative IgG4-associated AIH mimicking lymphoproliferative disorders.
Introduction
Autoimmune hepatitis (AIH) is an autoimmune liver disease characterized by hypergammaglobulinemia, the presence of serum autoimmune antibodies, and interface hepatitis. AIH is treated with immunosuppressants, including glucocorticoids, with or without azathioprine [1-3]. The diagnosis of AIH is usually made according to a scoring system, such as the Revised International Autoimmune Hepatitis Group (IAIHG) scoring system, due to heterogeneous clinical manifestations [4]. Recently, a new disease entity called Immunoglobulin G4 (IgG4)-associated AIH, which is different from classic AIH, has been described [5]. IgG4-associated AIH, as a subtype of AIH, is a rare disease characterized by the hepatic accumulation of IgG4-expressing plasma cells with markedly elevated serum IgG4 levels [6].
Differential diagnosis involves observation of markedly elevated serum IgG levels, presence of rouleaux formation in the peripheral smear, and low albumin/globulin ratios expressed in mono or polyclonal gammopathy in order to differentiate the condition from lymphoproliferative disorders such as multiple myeloma [7]. We report on a 47-year-old female with autoantibody-negative IgG4-related AIH that mimicked a lymphoproliferative disorder.
Case
All authors declare that written informed consent was obtained from the patient for publication of this case report and accompanying images. This study was approved by the Institutional Review Board of the Yeungnam University Hospital (IRB No: 2020-03-026).
A 47-year-old female visited our clinic for evaluation of abnormal liver function tests on a health-check examination. She had a previous history of a contrast allergy. She did not have any significant symptoms. There were no abnormal findings on physical examination. Abdominal ultrasound and non-contrast computed tomography revealed coarsened hepatic echotexture, without any biliary duct abnormalities, and no evidence of pancreatitis such as peripancreatic swelling or a sausage-like lesion (Fig. 1). Blood chemistry revealed the following: white blood cells, 3,650/μL; hemoglobin, 12 g/dL; platelets, 294×103/μL; serum total protein, 11.03 g/dL; serum albumin, 3.63 g/dL; total bilirubin, 1.02 mg/dL; serum aspartate aminotransferase (AST), 610 IU/L; alanine aminotransferase (ALT), 221 IU/L; alkaline phosphatase (ALP), 102 IU/L; gamma-glutamyl transferase (GGT), 176 IU/L; amylase, 65 IU/L; lipase, 54 IU/L; and prothrombin time-international normalized ratio, 1.12.
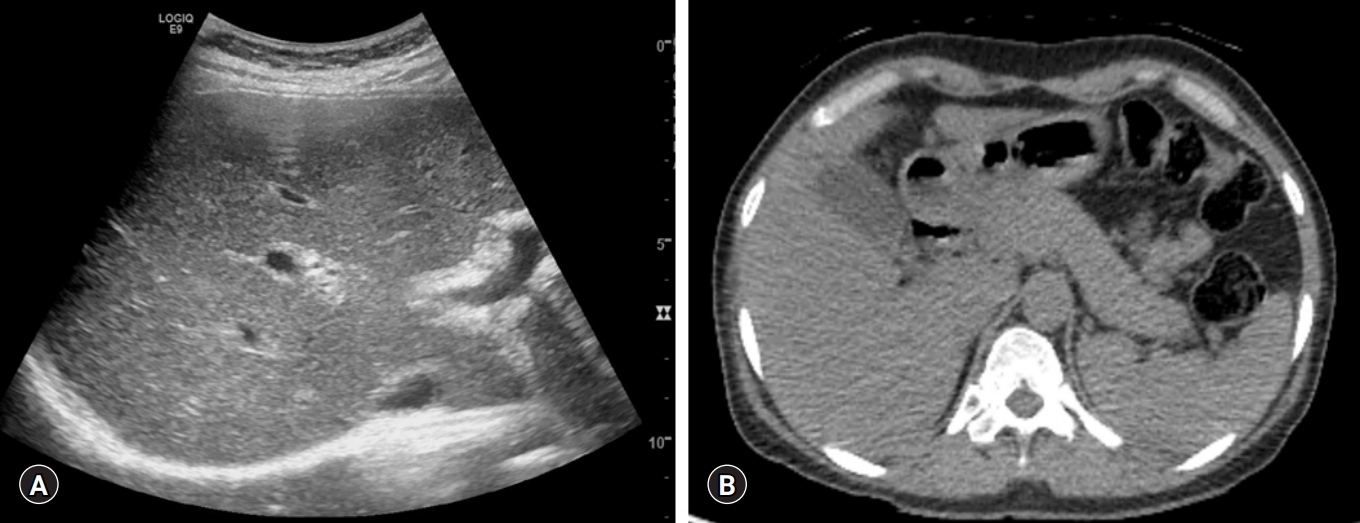
Abdominal sonography and computed tomography showing (A) increased echogenicity of the liver parenchyma with heterogeneous echotexture and (B) normal pancreas without significant evidence of autoimmune pancreatitis.
Additional serologic tests, including serum hepatitis B surface antigen, hepatitis C antibody, anti-nuclear antibody (ANA), anti-smooth muscle antibody (SMA), anti-mitochondria antibody, anti-neutrophil cytoplasmic antibody, anti-liver kidney microsomal type 1 antibody, and other viral tests—including Epstein-Barr virus and cytomegalovirus—were all negative. However, she exhibited a markedly elevated serum IgG level at 6,614 mg/dL (range, 700–1,600 mg/dL). Peripheral blood smear revealed the rouleaux formation of the red blood cells (RBC).
The patient was referred to the Department of Hematology to evaluate the possibility of a lymphoproliferative disorder, such as multiple myeloma (MM). Serum and urine protein electrophoresis, free light chain ratio, skeletal survey, and bone marrow aspiration were performed. All were normal with the exception of an elevated serum IgG4 subclass level, which was 221.3 mg/dL (range, 6–120 mg/dL).
A percutaneous ultrasound-guided liver biopsy was performed. The pathologic results revealed moderate spotty necrosis and interface hepatitis with highly elevated IgG4-positive plasma cells (10–15/high power field [HPF]), consistent with AIH (Fig. 2). The pretreatment revised IAIHG scoring was 17, consistent with a definite diagnosis of AIH. Detailed IAIHG scoring was as follows: histological feature, 4; no drug and alcohol history, 3; negative findings of viral markers, 3; hypergammaglobulinemia, 3; ALP/AST ratio <1.5, 2; and female, 2.
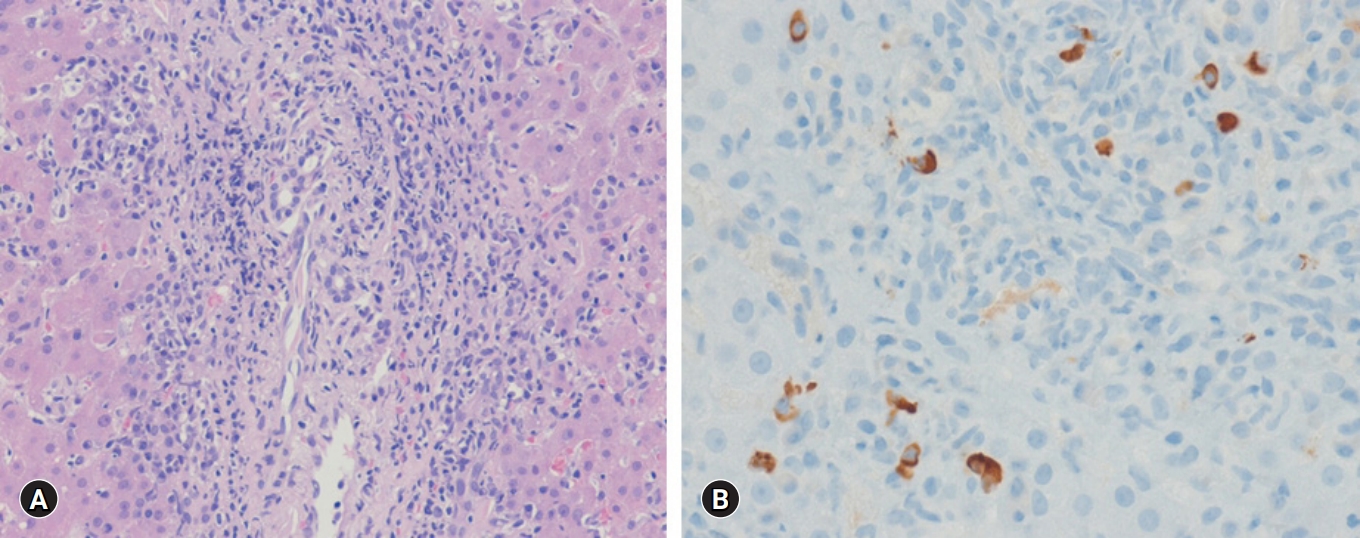
Histologic findings of liver. (A) Moderate interface hepatitis with lymphocytes and plasma cells infiltration in the portal tract is present (hematoxylin-eosin stain, ×200). (B) Immunoglobulin G4-positive plasma cells are seen (immunohistochemical stain, ×400).
Initially, she was treated with 60 mg prednisolone daily, which was tapered by 10 mg prednisolone weekly for 3 weeks. After 3 weeks, her laboratory test results were much improved as follows: AST, 25 IU/L; ALT, 23 IU/L; GGT, 67 IU/L; and IgG, 2,067 mg/dL. Subsequently, 50 mg azathioprine was added for maintenance treatment, and 30 mg prednisolone was tapered by 5 mg weekly for 4 weeks. After 4 weeks, her laboratory test results were slightly worsened as follows: AST, 57 IU/L; ALT, 65 IU/L; GGT, 103 IU/L; and IgG, 1,367 mg/dL. Considering the risk of possible worsening after early prednisone withdrawal, we increased the dose of prednisolone up to 20 mg and then slowly tapered every 2 weeks. After 1 month, all laboratory test results normalized (Fig. 3).
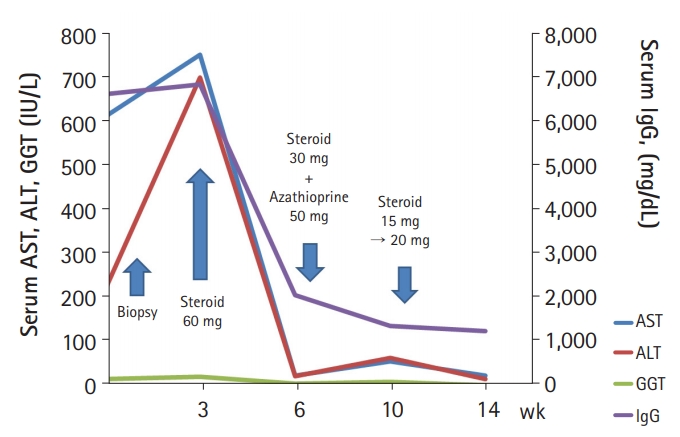
The clinical course of the patient. AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transferase; IgG, immunoglobulin G.
Discussion
In our case, the initial presumptive diagnosis was monoclonal or polyclonal gammopathy, such as MM or other lymphoproliferative diseases, based on the initial serological results including those for inverted albumin/globulin ratio (A/G) ratio (0.33), elevated serum IgG, rouleaux formation of RBC, and negative serologic autoimmune markers. However, based on the results of the liver biopsy and elevated serum IgG4 levels, the final diagnosis was IgG4-associated AIH which was treated with prednisolone and azathioprine.
Previous studies of IgG4-associated AIH are summarized in Table 1 [6,8-11]. Recently, a new disease entity called IgG4-associated AIH, which is different from classic AIH, has emerged [5]. Umemura et al. [5,6] had proposed that IgG4-AIH, as a subtype of AIH, is a rare disease characterized by the hepatic accumulation of IgG4-expressing plasma cells (≥10 IgG4 plasma cells per HPF) with markedly elevated serum IgG levels (≥135 mg/dL). Of the 60 patients with AIH in Japan, only two (3%) were diagnosed with IgG4-associated AIH [6]. The median values of IgG and IgG4 levels were 4,015 mg/dL and 560 mg/dL, respectively. These levels were higher than those of classic AIH (IgG, 2,940 mg/dL and IgG4, 22 mg/dL, respectively) [6]. Also, two patients were diagnosed with IgG4-associated AIH, with positive ANA and SMA (1 of 2 patients), which was defined as a subtype of AIH [6]. In a Western study, based on Umemura’s histologic criterion, the prevalence of IgG4-associated AIH was 25% (7 patients). The presence of the autoimmune antibodies was not significantly different from IgG4-associated AIH and classic AIH [8].

Autoantibody-negative immunoglobulin G4-associated autoimmune hepatitis
In our case, serum IgG and IgG4 levels were 6,614 mg/dL and 221.3 mg/dL, respectively. These markedly elevated IgG levels were four times greater than the normal upper level, which satisfied Umemura’s serologic criterion. In the liver, IgG4-bearing plasma cells of 10-15 HPF also met Umemura’s histologic criterion for diagnosis of IgG4-associated AIH. Owing to the initially all-negative autoimmune antibody serology, marked elevated hypergammaglobulinemia, and inverted A/G ratio in our case, we preferentially considered a lymphoproliferative disorder such as MM. Though most IgG4-associated AIH had positive ANA or SMA [6,8], the frequency of autoantibody-negative AIH in patients with acute and acute-severe clinical features reached 7% in one study [12]. Therefore, though patients show negative ANA, hypergammaglobulinemia, and elevated liver enzymes, additional serum IgG4 tests would be needed for differential diagnosis of IgG4-associated AIH. Then, when IgG4 is elevated, a subsequent liver biopsy is required for the definite diagnosis of IgG4-associated AIH, as with our case.
There can be some conflicts regarding IgG4-related disease (IgG4-RD) and IgG4-associated AIH, as noted by several studies [6,8-10]. The clinical features of IgG4-RD, which are similar to elevated IgG and IgG4 levels, include multiple organ involvement with storiform fibrosis. IgG4-RD preferentially affects the pancreas, bile duct, kidneys, and salivary glands, but rarely involves the liver [13,14]. However, it is still uncertain whether IgG4-associated AIH is a subtype of AIH or hepatic involvement of IgG4-RD. In our opinion, when evidence of multiple organ involvement and pathologic storiform fibrosis are not seen, the possibility of IgG4-associated AIH is higher than IgG4-RD.
Complete normalization of ALT and IgG levels is the clinical target for the treatment of AIH [15]. The treatment of IgG4-AIH is not much different from that of AIH, and involves glucocorticoids with or without azathioprine [16]. The efficacy of these treatments could be predicted by the accumulation of plasma cells in the liver [16]. However, some studies reported that the time to return to ALT normalization after administration of glucocorticoids is shorter in patients with IgG4-associated AIH than in those with classic AIH (3.7 months vs. 6.7 months, respectively) [10,11]. In our study, mild flare-up while switching from a glucocorticoid to combination therapy was observed, which may have resulted from rapid tapering of the glucocorticoid dose by 5 mg weekly. The early withdrawal of immunosuppressants could be a risk factor for flares of AIH, which require increased doses of glucocorticoids [15]. Therefore, monitoring intervals should be adjusted according to the individual clinical response, which could be extended to 1–3 months with combination treatment [15].
In conclusion, IgG4-associated AIH should be contemplated in patients with marked hypergammaglobulinemia and abnormal liver function test results, even when the autoimmune test is negative. Liver biopsy should be required for the diagnosis of IgG4-associated AIH in these patients with elevated serum IgG4.
Notes
Conflicts of interest
Joon Hyuk Choi serves as an editor-in-chief of the Yeungnam University Journal of Medicine, but has no role in the decision to publish this article. Except for that, no potential conflict of interest relevant to this article was reported.
Author contributions
Conceptualization: MKK, JGP, JHC; Data curation: MKK; Methodology: MKK, JGP; Investigation: MKK, JHC; Resources: JHC; Project administration: JHC; Visualization: JHC; Writing-original draft: MKK; Writing-review & editing: JGP.