Estrogen-secreting adrenocortical carcinoma
Article information

Abstract
Adrenocortical carcinoma is a rare type of endocrine malignancy with an annual incidence of approximately 1–2 cases per million. The majority of these tumors secrete cortisol, and a few secrete aldosterone or androgen. Estrogen-secreting adrenocortical carcinomas are extremely rare, irrespective of the secretion status of other adrenocortical hormones. Here, we report the case of a 53-year-old man with a cortisol and estrogen-secreting adrenocortical carcinoma. The patient presented with gynecomastia and abdominal discomfort. Radiological assessment revealed a tumor measuring 21×15.3×12 cm localized to the retroperitoneum. A hormonal evaluation revealed increased levels of estradiol, dehydroepiandrosterone sulfate, and cortisol. The patient underwent a right adrenalectomy, and the pathological examination revealed an adrenocortical carcinoma with a Weiss’ score of 6. After surgery, he was treated with adjuvant radiotherapy. Twenty-one months after treatment, the patient remains alive with no evidence of recurrence.
Introduction
Adrenocortical carcinoma is a very rare type of tumor reported to occur in approximately 1–2 per million people per year [1,2]. Adrenocortical carcinomas can be divided into functional or nonfunctional tumors according to the hormone secretion status; however, most adrenocortical tumors secrete cortisol and rarely secrete androgens and aldosterone [3,4]. International reports have rarely described adrenocortical carcinomas that secrete estrogen; although a few existing reports have used the term feminizing adrenal tumors (FATs) to indicate adrenocortical carcinomas that secrete estrogen in men, such cases have not previously been reported in Korea [5]. We report a case of estrogen-secreting adrenocortical carcinoma in a 53-year-old man who presented with abdominal discomfort and gynecomastia.
Case
Patient: A 53-year-old man.
Chief complaint: Gynecomastia and abdominal discomfort.
Present illness: A 53-year-old man with a history of hypertension visited the Department of Family Medicine at our hospital with the complaint of a 6-month history of bilateral breast enlargement, as well as right-sided flank pain and abdominal discomfort. After abdominal computed tomography (CT) revealed an abdominal mass, he was referred to an endocrinologist for a hormone assessment and further examination.
Past history: He had been diagnosed with hypertension 5 years earlier and was taking medication.
Physical examination: The patient's height and weight were 173 cm and 72 kg, and his body weight had increased by approximately 10 kg over the last 6 months. His blood pressure was 120/80 mmHg, body temperature was 36.5ºC, pulse rate was 68 beats/minute, and respiration rate was 18 breaths/minute. His consciousness was clear, and he had no symptoms such as palpitation, sweating, or headache. His conjunctiva and sclera were normal, and a head and neck examination detected no moon face, flushing, or other specific symptoms. During a chest examination, a 4-cm-sized gynecomastia was observed bilaterally with tenderness. The patient's respiratory sounds were normal, and no heart murmurs were detected. An abdominal examination revealed no purple striae but did reveal tenderness in the right upper quadrant and a palpable hard, fixed mass with an approximate size of 15 cm. The patient's bowel sounds were normal, and he exhibited no edema or tenderness in his limbs. There were no skin rashes or specific findings of the joints. A lower limb test revealed normal sensory and motor functions. The patient denied erectile dysfunction and reported a normal shaving frequency of once every 2 days.
Laboratory findings: A complete blood cell count during blood analysis revealed a leukocyte count of 9,330/mm³ (neutrophils, 75.3%), hemoglobin level of 11.4 g/dL, and platelet count of 329,000/mm³. A comprehensive metabolic panel indicated the following levels: aspartate aminotransferase and alanine aminotransferase, 31 and 23 IU/L, respectively; alkaline phosphatase, 80 IU/L; total protein, 6.2 g/dL; albumin, 3.3 g/dL; calcium, 8.8 mg/dL; blood urea nitrogen, 9.0 mg/dL; and creatinine, 0.7 mg/dL. A hormonal assessment revealed increases in the levels of dehydroepiandrosterone, estradiol, and 24-hour urine cortisol but decreases in the levels of follicle-stimulating hormone, testosterone, and adrenocorticotropic hormone. The patient's serum cortisol and luteinizing hormone levels were normal (Table 1). His morning plasma cortisol level was 16.3 μg/dL (range, <5 μg/dL), and this was not suppressed during an overnight dexamethasone suppression test. The patient rejected an additional planned low-dose dexamethasone suppression test. A 24-hour urine test yielded normal vanillylmandelic acid, metanephrine, and epinephrine levels.

Hormone study
Radiological findings: Simple chest radiography yielded normal findings. Abdominal CT showed a heterogeneously well-defined single mass with dimensions of 18×11×18 cm between the liver and right kidney (Fig. 1). On renal CT, the mass was not observed to have infiltrated the liver, kidney, and surrounding blood vessels, and lymph node metastasis and ascites were not observed. A chest CT scan and bone scan showed no metastasis.

Contrast-enhanced abdominal computed tomography reveals a right adrenal mass. Axial (A) and coronal (B) images of a well-defined, heterogeneously enhancing mass measuring 18×11×18 cm between the liver and right kidney.
Treatment and progress: The patient underwent open surgery under general anesthesia to remove the right adrenal tumor. The tumor was found to have adhered to the liver, kidney, and inferior vena cava, and a partial liver resection was performed. Inferior vena cava and kidney involvement were not observed. There were no postoperative complications. The final tumor classification was pT2N0M0, stage 2, and the patient received additional adjuvant radiation therapy. Postoperatively, he was given replacement therapy with hydrocortisone for suspected adrenal insufficiency. Three months after surgery, his estradiol level was 70.32 pg/mL and the gynecomastia had almost disappeared. The patient remains in good health without any local recurrence or metastasis at 21 months after diagnosis and treatment, and the gynecomastia has almost resolved.
Pathological findings: A gross pathology examination of the surgically resected tumor revealed a size of 21×15.3×12 cm and a smooth and partially fibrous outer surface. An analysis of the tumor cross-section revealed encapsulated yellowish-brown nodules and widely distributed necrotic and hemorrhagic lesions (Fig. 2).
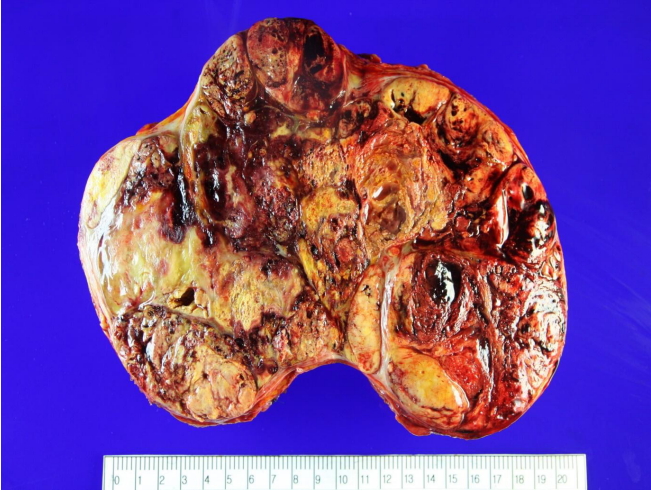
Gross appearance of the resected adrenal tumor. The mass was large, solitary, and circumscribed tumor (21×15.3×12 cm). The cut section is yellowish-tan in color, with a variegated appearance. Many areas of necrosis and hemorrhage are visible.
A microscopic examination revealed tumor necrosis and readily apparent mitosis. The histopathologic examination revealed necrosis, diffuse architecture, a high nuclear grade, high mitotic rate, atypical mitotic figures, and a clear cell frequency of <25% (Fig. 3). The immunohistochemistry results included positive staining for inhibin α, MART-1, and calretinin and a Ki67 proliferation index of 20% (Fig. 4). No tumor invasion was observed in the resected mass.
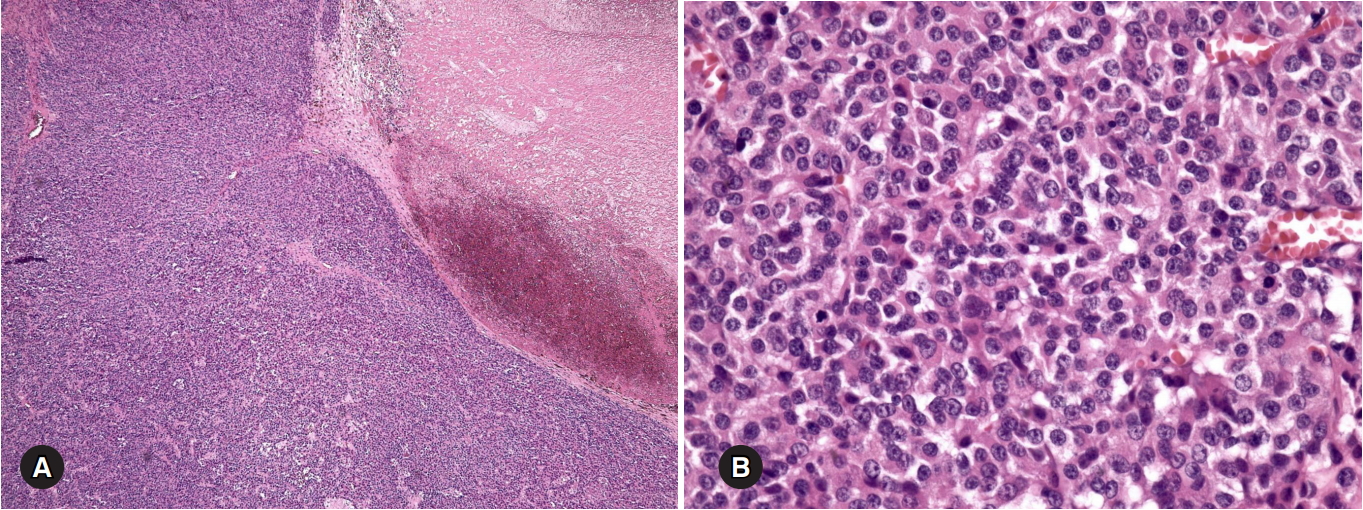
Microscopic tumor findings. (A) The tumor exhibits a diffuse solid growth pattern with necrosis on the right side (hematoxylin and eosin [H&E] stain, ×40). (B) The tumor cells are round-to-oval in shape with high-nuclear-grade nucleoli. Mitoses are frequently observed (H&E stain, ×400).
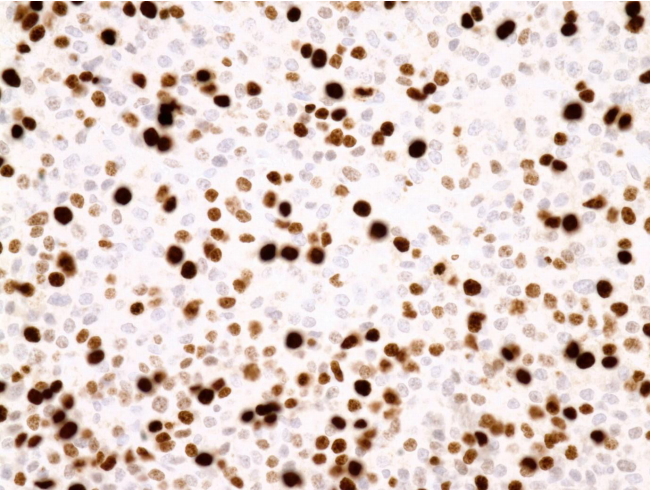
Immunohistochemical staining of the tumor. The Ki-67 index of the tumor cells is 20% (×400).
Discussion
Feminizing adrenal carcinomas are very rare, accounting for 1–2% of all adrenocortical carcinomas [5]. According to one study, fewer than 100 cases were reported prior to 1994, and only a few cases have since been reported. Additionally, no previous report has described a case of a FAT in Korea [6].
Clinical manifestations of FATs include gynecomastia, testicular atrophy, and a loss of sexual desire; we note that our patient complained of gynecomastia and loss of libido [7]. Although gynecomastia often occurs in men as a consequence of adverse drug reactions, renal failure, and cirrhosis [8], it is very rarely is caused by the direct secretion of estrogen from an adrenocortical carcinoma.
In our patient, we observed no tumor involvement in the surrounding organs or metastasis. However, functional adrenocortical carcinomas are frequently malignant, especially if estrogen is secreted [9-11]. Adrenocortical carcinoma is diagnosed through gross examination, microscopic examination, and immunohistochemical evaluation. The Weiss score is used as a histologic criterion, and a diagnosis of malignancy is made if at least three of nine criteria are met. In our case, six of nine criteria were satisfied, and the patient was diagnosed with a malignant tumor [12] (Table 2).

Weiss score
Adrenocortical carcinoma is an aggressive tumor with a very poor prognosis. A study conducted at the Memorial Sloan Kettering Cancer Center reported 5-year survival rates of 60% for stage 1–2 adrenocortical carcinomas and 10% for stage 3–4 cases [13]. No statistical analyses of data regarding the prognosis of FAT have been reported; however, only two patients have survived for more than 10 years after diagnosis, irrespective of treatment [5]. Among cases of adrenocortical carcinoma, prognostic factors include age, mitotic count and the Ki-67 index. However, staging and the possibility of surgical resection are the most important factors [14-16]. Although locally advanced stage 1–3 adrenocortical carcinomas can be cured by surgical resection, the possibility of local recurrence and metastasis within 2 years is approximately 65% among patients who underwent a radical resection [17]. Laparoscopic adrenalectomy is considered safe, but open surgery is preferred as a curative treatment option. In our case, the patient underwent a radical resection via open surgery, which addressed the large tumor size and the adhesions to the liver, kidney, and inferior vena cava [18,19].
According to the National Comprehensive Cancer Network guideline, the delivery of external radiation therapy to the tumor removal site is recommended if the case is considered to be at a high risk of local recurrence after radical resection. In our case, the massive tumor size and Ki-67 index of 20% indicated a high-risk status, and surgical resection was followed by radiotherapy. Previous studies have reported that radiotherapy could significantly reduce the recurrence rate in cases of adrenocortical carcinoma [20]; however, FATs are rare and the evidence regarding treatment remains at a case-centered level.
In conclusion, we diagnosed an adrenocortical carcinoma with estrogen secretion in a male patient with gynecomastia, which was treated via surgery and radiation therapy and monitored through continuous follow-up visits. As noted, FATs are rarely reported in the international literature and have not previously been reported in Korea, and therefore, we have presented our case in the context of a literature review.
Notes
No potential conflicts of interest relevant to this article were reported.