E-Submission
E-SubmissionPubMed Central, CAS, DOAJ, KCI

Articles
- Page Path
- HOME > J Yeungnam Med Sci > Volume 40(Suppl); 2023 > Article
-
Case report
A rare pathogenic variant identified in a heart transplant recipient with hereditary transthyretin amyloidosis: a case report -
Myeong Seop Kim
 , Soo Youn Lee, Kyung-Hee Kim
, Soo Youn Lee, Kyung-Hee Kim -
Journal of Yeungnam Medical Science 2023;40(Suppl):S98-S104.
DOI: https://doi.org/10.12701/jyms.2023.00241
Published online: May 30, 2023
Division of Cardiology, Department of Internal Medicine, Cardiovascular Center, Incheon Sejong Hospital, Incheon, Korea
- Corresponding author: Kyung-Hee Kim, MD, PhD Division of Cardiology, Department of Internal Medicine, Cardiovascular Center, Incheon Sejong Hospital, 20 Gyeyangmunhwa-ro, Gyeyang-gu, Incheon 21080, Korea Tel: +82-32-240-8809 • Fax: +82-32-240-8809 • E-mail: learnbyliving9@gmail.com
Copyright © 2023 Yeungnam University College of Medicine, Yeungnam University Institute of Medical Science
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 996 Views
- 37 Download
Abstract
- Hereditary transthyretin (ATTRv) amyloidosis is a rare and complex genetic disorder that can lead to life-threatening cardiac amyloidosis and rapid disease progression. Early diagnosis and treatment with disease-modifying drugs can improve patient outcomes; however, heart transplantation may be necessary in some patients. We present the unique case of a 65-year-old Korean woman diagnosed with ATTRv amyloidosis after experiencing progressive neurological symptoms, followed by heart failure. Despite the absence of significant symptoms of heart failure, subsequent screening revealed cardiac amyloid infiltration, which caused left ventricular hypertrophy and rapid disease progression. The patient underwent successful heart transplantation, and subsequent genetic testing revealed a pathogenic variant, NM_000371.3:c.425T>C (p.Val142Ala), which affects both the nerves and heart and has not been previously reported in Korea. Our report underscores the potential benefits of heart transplantation in managing advanced ATTRv amyloidosis and emphasizes the need for continued research on the genetic heterogeneity of the disease. Clinicians should consider ATTRv amyloidosis in the differential diagnosis of patients presenting with neurological symptoms and heart failure, particularly in those with a family history of the disease.
- Hereditary transthyretin (ATTRv) amyloidosis is a rare and complex genetic disorder that can result in a wide range of debilitating symptoms and complications [1]. Although it can be challenging to diagnose and manage, recent advances in available therapies have shown promise in slowing or preventing disease progression and improving patient outcomes. In this case report, we present the unique and compelling case of a 65-year-old Korean woman diagnosed with ATTRv amyloidosis after experiencing progressive neurological symptoms and heart failure. Through timely intervention and careful management, the patient underwent life-saving heart transplantation, demonstrating the potential benefits of early detection and treatment for managing ATTRv amyloidosis. Furthermore, this case highlights a rare pathogenic variant, NM_000371.3:c.425T>C (p.Val142Ala), which has not been previously reported in Korea.
Introduction
- Ethical statements: This study was approved by the Institutional Review Board (IRB) of Incheon Sejong Hospital (IRB No: 2023-03-003), and informed consent was obtained from the patient.
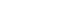
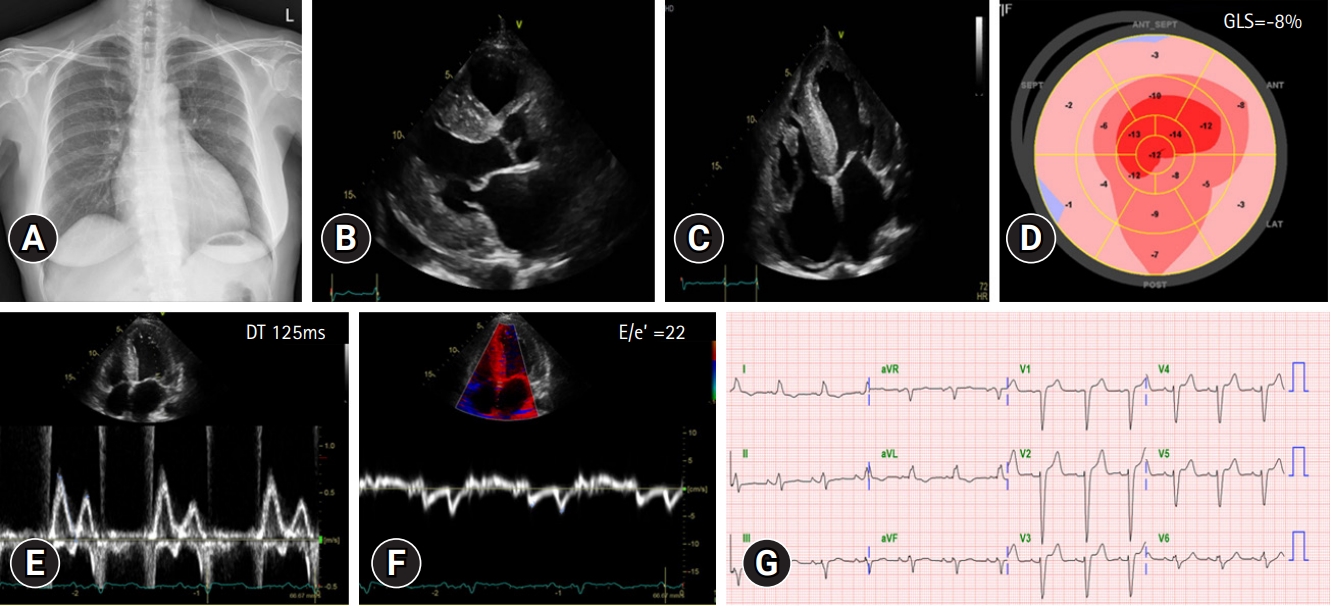
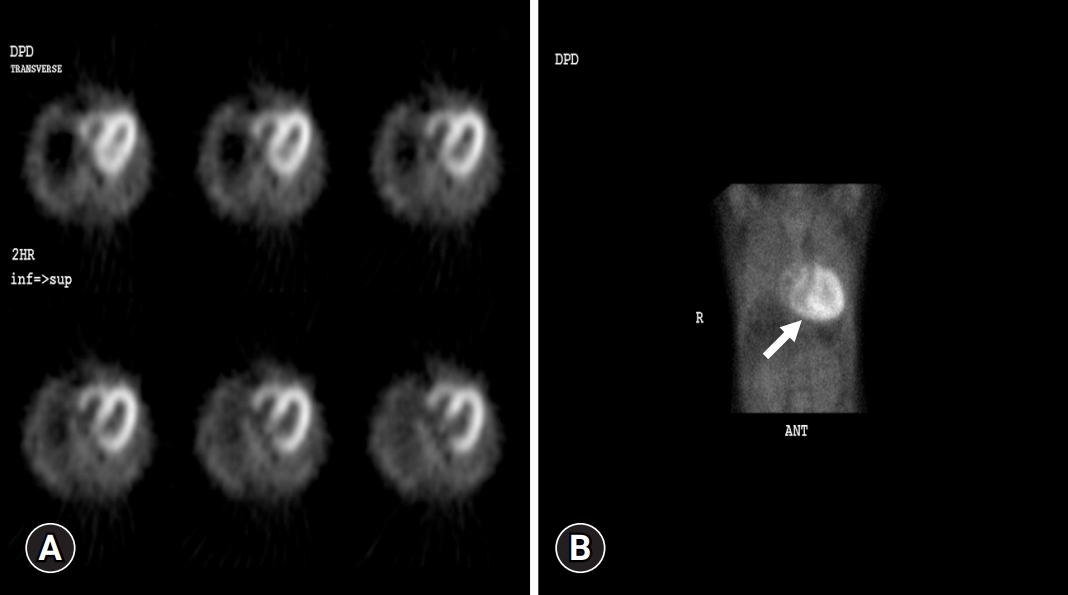
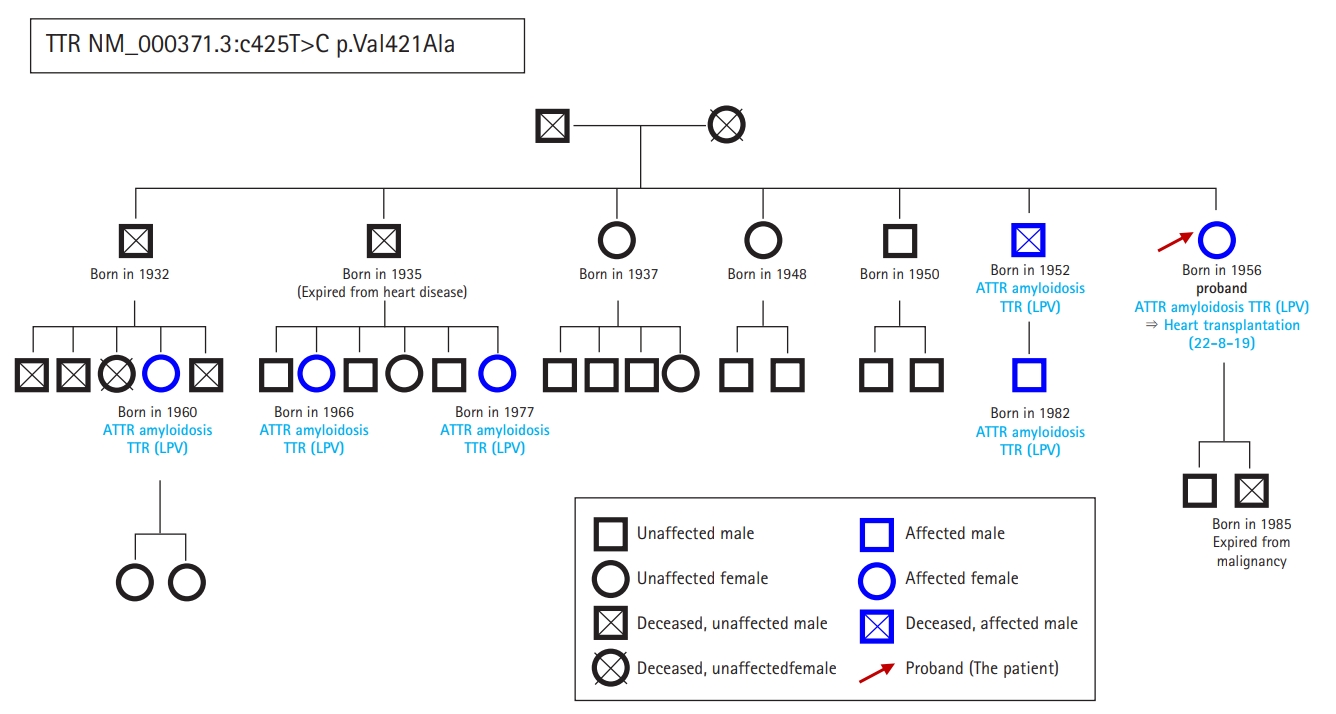
- In September 2021, a 65-year-old Korean female patient presented to our hospital for genetic testing and cardiac screening because of a family history of unknown heart disease and her older brother’s recent diagnosis of ATTRv amyloidosis. The patient had no symptoms of heart failure at the time of presentation but reported cardiomegaly on chest radiography (Fig. 1A). Two of the patient’s seven siblings died of unknown heart disease, and the patient had an older brother who was diagnosed with ATTRv amyloidosis in 2018 and was being treated for heart failure, including with tafamidis. The patient complained of numbness in the dorsa of the feet and dizziness when standing up. The patient had a surgical history of carpal tunnel syndrome in both hands and had not taken any medication. Neurological examination revealed that the pain and vibration sensation in the lower extremities had decreased, and deep tendon reflexes in the ankle were absent. No specific findings were identified in the ophthalmic examinations. Electrocardiography (ECG) revealed sinus rhythm, left bundle branch block, and low QRS voltage; however, the Q waves in leads II, III, and aVF, which had been confirmed in her brother’s ECG, were absent (Fig. 1G). A 24-hour ambulatory ECG monitoring showed multiple premature ventricular beats. Transthoracic echocardiography (TTE) revealed left ventricular (LV) wall thickness of 15 mm, LV ejection fraction of 29%, and diastolic dysfunction (Fig. 1B–1F). In addition, TTE showed reduced longitudinal strain with an apical sparing pattern (Fig. 1D). The monoclonal gammopathy pattern on serum, urine protein electrophoresis, and immunofixation was not identified and the free kappa/lambda level was normal. Blood tests confirmed normal values, except for an increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) level of 3,055 pg/mL and high-sensitivity troponin I level of 0.219 ng/mL. Coronary angiography revealed normal coronary arteries. A nerve conduction study confirmed findings compatible with carpal tunnel syndrome in both hands and sensory abnormalities in the lower extremities, including the sural nerve (Fig. 2). Orthostatic hypotension was demonstrated by blood pressure monitoring using the tilt table test. Technetium-99m-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy revealed amyloid deposition in both ventricles (Fig. 3). Finally, cardiac biopsy was performed via cardiac catheterization, and the biopsy specimen was removed from the right ventricle. The biopsy findings demonstrated accumulation of amyloid fibrils in the myocardium (Fig. 4). Genetic testing using a blood sample revealed the presence of a transthyretin (TTR) pathogenic variant, NM_000371.3:c.425T>C (p.Val142Ala), which was confirmed in her older brother (Fig. 5). Optimal medical therapy was initiated, including heart failure medications and tafamidis. As the patient’s vital signs were stable without any further symptoms, treatment follow-up occurred in the outpatient clinic. In April 2022, the shortness of breath experienced by the patient worsened, warranting New York Heart Association class III functional classification, and her LV ejection fraction did not improve as determined by TTE. Therefore, cardiac resynchronization therapy was performed. However, serial images obtained through TTE in the outpatient clinic showed no signs of improvement thereafter. Her NT-proBNP level and right ventricular systolic pressure continued to increase, necessitating heart transplantation. Additional tests were performed to investigate the potential involvement of other organ systems, including renal function tests, central neurological studies, liver enzyme tests, pulmonary function tests, and computed tomography. No specific findings were observed on these tests. Routine esophagogastroduodenoscopy and colonoscopy with random biopsies revealed normal findings. In August 2022, the patient received an organ from a deceased donor and underwent orthotopic heart transplantation at our hospital. Subsequent TTE revealed normal heart function. The patient was discharged after a favorable postoperative course. Six months after the heart transplantation, a cardiac biopsy was performed on the patient, which revealed no amyloid deposits or signs of rejection. The patient is currently maintaining the course of tafamidis and immunosuppressants and is doing well without any shortness of breath or any additional neurological symptoms. The patient’s pedigree was confirmed (Fig. 6).
Case
- Cardiac amyloidosis is a group of rare and complex disorders characterized by the deposition of amyloid proteins in the heart, which can lead to progressive cardiomyopathy and heart failure. There are three main types of cardiac amyloidosis, primary (AL), secondary (AA), and hereditary (ATTRv), each with a unique pathogenesis and clinical presentation [2]. Among these, hereditary ATTRv amyloidosis is caused by genetic mutations in the TTR gene and is associated with more than two-thirds of the pathogenic variants of TTR [3]. In this form, the misfolded amyloid protein is deposited not only in the heart but also in the nerves, kidneys, and eyes, leading to a wide range of debilitating symptoms and complications. Therapeutic agents, including TTR tetramer stabilizers (such as tafamidis) and gene-silencing treatments, have been developed and their effects have been demonstrated in patients with hereditary ATTRv amyloidosis, in whom the heart and nerves are involved [4]. Disease progression is delayed in patients diagnosed and treated in early stages, and these patients have a reduced mortality risk compared with untreated patients [5,6]. Despite the availability of treatment, early diagnosis and genetic testing are crucial for effective disease management and improved patient outcomes. Patients with advanced disease may not respond to these drugs, leading to disease progression and the need for heart transplantation. Unfortunately, the availability of deceased donors for transplantation is limited and the waiting time is long, making transplantation challenging.
- In the case of hereditary ATTRv amyloidosis, where symptoms usually appear in adult patients, early evaluation of young family members with molecular genetic testing of blood samples and subsequent treatment can improve their prognosis [5]. Different TTR mutations causing hereditary ATTRv amyloidosis have been reported in various races and ethnicities worldwide [1,7]. Although the p.Val142Ile variant has a higher prevalence among African American populations, it is not the only TTR mutation that causes cardiac amyloidosis in this population; other mutations may be more prevalent in other populations [8]. The THAOS (Transthyretin Amyloidosis Outcomes Survey) study, a global registry of patients with TTR amyloidosis, has demonstrated that different TTR mutations causing hereditary ATTRv amyloidosis are present in various races and ethnicities worldwide, including in Europe and the Americas. Therefore, clinicians should consider the possibility of TTR amyloidosis in patients presenting with symptoms of cardiomyopathy, particularly in those with a family history of the disease or a known family history of TTR mutations.
- The frequency of TTR-related cardiac amyloidosis in the Korean population is not as well-known as in Western countries. In this case report, we present a unique case of a Korean patient with a previously unreported pathogenic variant, NM_000371.3:c.425T>C (p.Val142Ala), which affected both the nerves and heart as a mixed type. The patient survived after undergoing heart transplantation. This case highlights the importance of early diagnosis and genetic testing for ATTRv amyloidosis, particularly in families with a history of neurological symptoms or heart failure. The identification of a new pathogenic variant in this case underscores the need for continued research on the genetic diversity of ATTRv amyloidosis. In addition, this case report suggests that genetic testing in clinically suspected patients may be helpful for early diagnosis and treatment, ultimately leading to better patient outcomes. Finally, this report contributes to the growing body of knowledge on ATTRv amyloidosis and emphasizes the importance of continued research, genetic testing, and clinical management of individuals living with this complex disorder.
- In conclusion, genetic testing of patients with suspected hereditary ATTRv amyloidosis may aid in early diagnosis and treatment, ultimately improving their prognosis. This case report emphasizes the importance of genetic testing for ATTRv amyloidosis, particularly in families with a history of neurological symptoms or heart failure. Furthermore, the identification of a new pathogenic variant in this case highlights the need for continued research on the genetic diversity of ATTRv amyloidosis.
Discussion
-
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
-
Acknowledgments
The authors thank the patient for her consent to publish this case report.
-
Funding
None.
-
Author contributions
Conceptualization, Formal analysis: all authors; Methodology: MSK, KHK; Investigation, Visualization: KHK; Writing-original draft: MSK; Writing-review & editing: SYL, KHK.
Notes


- 1. Damy T, Kristen AV, Suhr OB, Maurer MS, Planté-Bordeneuve V, Yu CR, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J 2019;43:391–400.ArticlePubMedPMCPDF
- 2. Rapezzi C, Aimo A, Serenelli M, Barison A, Vergaro G, Passino C, et al. Critical comparison of documents from scientific societies on cardiac amyloidosis: JACC State-of-the-Art Review. J Am Coll Cardiol 2022;79:1288–303.ArticlePubMed
- 3. Lioncino M, Monda E, Palmiero G, Caiazza M, Vetrano E, Rubino M, et al. Cardiovascular involvement in transthyretin cardiac amyloidosis. Heart Fail Clin 2022;18:73–87.ArticlePubMed
- 4. Griffin JM, Rosenthal JL, Grodin JL, Maurer MS, Grogan M, Cheng RK. ATTR amyloidosis: current and emerging management strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol 2021;3:488–505.ArticlePubMedPMC
- 5. Coelho T, Conceição I, Waddington-Cruz M, Keohane D, Sultan MB, Chapman D, et al. A natural history analysis of asymptomatic TTR gene carriers as they develop symptomatic transthyretin amyloidosis in the Transthyretin Amyloidosis Outcomes Survey (THAOS). Amyloid 2022;29:228–36.ArticlePubMed
- 6. Ghoneem A, Bhatti AW, Khadke S, Mitchell J, Liu J, Zhang K, et al. Real-world efficacy of tafamidis in patients with transthyretin amyloidosis and heart failure. Curr Probl Cardiol 2023;48:101667.ArticlePubMed
- 7. Dispenzieri A, Coelho T, Conceição I, Waddington-Cruz M, Wixner J, Kristen AV, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis 2022;17:236.ArticlePubMedPMC
- 8. Chandrashekar P, Alhuneafat L, Mannello M, Al-Rashdan L, Kim MM, Dungu J, et al. Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: a systematic review. Circ Genom Precis Med 2021;14:e003356.ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite